
Abstract
Reactive oxygen species (ROS) are implicated in reperfusion injury after focal cerebral ischemia (FCI). Reactive oxygen species regulate activity of transcription factors like NF-κB. The authors investigated the role of ROS in NF-κB activity after FCI using transgenic mice that overexpressed human copper/zinc-superoxide dismutase (SOD1) and that had reduced infarction volume after FCI. Superoxide dismutase transgenic and wild-type mice were subjected to 1 hour of middle cerebral artery occlusion (MCAO) and subsequent reperfusion. Immunohistochemistry showed SOD1 overexpression attenuated ischemia-induced NF-κB p65 immunoreactivity. Colocalization of NF-κB and the neuronal marker, micro-tubule-associated proteins (MAPs), showed that NF-κB was up-regulated in neurons after FCI. Electrophoretic mobility shift assays showed that SOD1 overexpression reduced ischemia-induced NF-κB DNA binding activity. Supershift assays showed that DNA–protein complexes contained p65 and p50 subunits. Immunoreactivity of c-myc, an NF-κB downstream gene, was increased in the ischemic cortex and colocalized with NF-κB. Western blotting showed that SOD1 overexpression reduced NF-κB and c-Myc protein levels in the ischemic brain. Colocalization of c-Myc and TUNEL staining was observed 24 hours after FCI. The current findings provide the first evidence that SOD1 overexpression attenuates activation of NF-κB after transient FCI in mice and that preventing this early activation may block expression of downstream deleterious genes like c-myc, thereby reducing ischemic damage.
Reactive oxygen species (ROS) have been implicated in the pathogenesis of ischemic brain injury (Kontos, 1985; Siesjö et al., 1989). When ROS are overproduced during postischemic reperfusion, they exceed the capacity of antioxidant enzymes like superoxide dismutases (SODs), thereby causing oxidative stress, which damages cells. Reactive oxygen species are involved in reperfusion after cerebral ischemia (Chan, 1996). Overexpression of copper/zinc-SOD (SOD1) in transgenic (Tg) mice or rats is neuroprotective in brain injuries such as cold-induced brain edema (Chan et al., 1991), traumatic brain injury (Mikawa et al., 1996), global ischemia (Chan et al., 1998; Murakami et al., 1997), and transient focal cerebral ischemia (FCI) (Yang et al., 1994). In SOD1 Tg mice, brain edema, infarct volume, and DNA fragmentation are significantly reduced after transient FCI (Fujimura et al., 1999b; Kamii et al., 1995; Kinouchi et al., 1991), whereas they are markedly increased in SOD1 knockout mutant mice (Kondo et al., 1997), suggesting that superoxide radicals are involved in the pathogenesis of neuronal cell death after transient FCI. However, the molecular mechanisms underlying regulation of neuronal protection in SOD1 Tg animals are not clear.
Reactive oxygen species play a role in the cellular processes, including regulation of transcription factor activity. Nuclear factor-κB (NF-κB) responds to oxidative stress induced by ROS (Schreck et al., 1991). On activation, NF-κB dissociates from the inhibitory protein IκB in the cytoplasm, translocates into the nucleus, and binds to its cognate sequence of target genes. NF-κB can be composed of hetero- or homodimers of proteins such as p50 and p65.
NF-κB is activated by a variety of stimuli that induce ROS, and may play a role in the pathogenesis of oxidative stress-associated acute injuries and neurodegenerative disorders, including focal ischemia. However, the role of ROS in activating NF-κB after focal ischemia is not clear. Because of the apparent link between ROS and NF-κB, the authors were prompted to determine whether alteration of NF-κB activity could contribute to SOD1 neuronal protection against transient ischemic insult. On the other hand, NF-κB has been implicated in the regulation of apoptosis (Foo and Nolan, 1999). Evidence of its dual roles (that is, antiapoptotic versus proapoptotic) has caused controversy (Baichwal and Baeuerle, 1997; Lipton, 1997). A recent report demonstrated that NF-κB may play a proapoptotic role in transient FCI (Schneider et al., 1999). However, the exact downstream effectors of the mechanism by which NF-κB promotes apoptosis are poorly defined. Several genes induced by ischemia and reperfusion contain NF-κB binding sites, including c-myc (Nakagomi et al., 1996), an apoptotic inducer (Evan et al., 1992). Two functional NF-κB sites have been identified within the c-myc promoter, which is transactivated by NF-κB (Duyao et al., 1990; La Rosa et al., 1994). Moreover, NF-κB activation is required for the expression of c-Myc (Romashkova and Makarov, 1999). Thus, the authors were prompted to determine whether c-Myc is involved in cell death pathways after transient FCI. The authors' aim was to investigate the role of ROS in NF-κB activation after FCI, using SOD1 Tg and wild-type mice that were subjected to ischemia and reperfusion. In addition, to address the putative mechanisms involved, the authors evaluated c-Myc activation after FCI.
MATERIALS AND METHODS
Focal cerebral ischemia
Experiments were performed in accordance with National Institutes of Health guidelines and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Transgenic mice of the TgHS/SF-218 strain, which carries the human copper/zinc-SOD gene, were derived from the founder stock (Epstein et al., 1987). There were no observable phenotypic differences between the Tg mice and their wild-type normal littermates. Transgenic mice (3-month-old males, 35 to 40 g) with a threefold overexpression in SOD1 activity in brain cells (Epstein et al., 1987) and wild-type mice were subjected to focal cerebral ischemia and reperfusion in a randomized, blind manner. Focal ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO) with a nylon monofilament suture as described previously (Yang et al., 1994). Mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a facemask. Rectal temperature was controlled at 37°C with a homeothermic blanket. The left femoral artery was cannulated for measurement of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. Blood gas was analyzed with a pH/Blood Gas Analyzer (Chiron Diagnostics Ltd., Essex, U.K.). The left common carotid artery was exposed and the left external carotid artery and its branches were electrocoagulated. An 11.0-mm 5–0 surgical monofilament nylon suture, blunted at the tip, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 minutes of proximal MCAO, blood flow was restored by removing the suture. Control normal wild-type mice did not undergo surgery. After 60 minutes of MCAO, the mice were allowed to recover for 5 minutes and 1, 4, and 24 hours. Physiologic parameters were monitored throughout the studies and values were the same as previously reported (Fujimura et al., 1999a).
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L phosphate-buffered saline (PBS), pH 7.4, 5 minutes and 1, 4, and 24 hours after ischemia. Brains were removed, postfixed for 12 hours in 4% formaldehyde, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. Endogenous peroxidase activity was quenched by immersing the sections in 3% H2O2 for 10 minutes. A nonspecific blocking procedure, using 1% bovine serum albumin, was performed before application of primary antibodies. Then the sections were incubated with NF-κB or c-Myc polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) at a dilution of 1:200 for 72 hours or overnight at 4°C. The sections were incubated with avidin-biotin-horseradish peroxidase (ABC kit; Vector Laboratories, Burlingame, CA, U.S.A.) to localize the primary antibodies. A diaminobenzidine (DAB) substrate was used for visualization of the catalyzed peroxidase-reaction product. Then the nuclei were counterstained with methyl green solution for 10 minutes and mounted. For double-labeling studies, the neuron-specific marker, MAPs (Sigma, St. Louis, MO, U.S.A.), was used to identify the phenotype of the NF-κB– or c-Myc–expressing cells at a 1:500 dilution for 5 hours at room temperature. Sections were incubated with the ABC kit and visualized with DAB/nickel sulfate. As a negative control, sections were incubated without primary antibodies.
Fluorescence double-labeling studies of NF-κB and c-Myc
Brain sections were prepared as described above. A nonspecific blocking procedure, using 20% horse serum albumin, was performed before application of primary antibodies. Then the sections were incubated with NF-κB p65 goat polyclonal antibodies (Santa Cruz Biotechnology) at a dilution of 1:150 for 72 hours at 4°C. The sections were incubated with secondary biotinylated horse anti-goat immunoglobulin G and then with Fluorescein Avidin DCS (Vector) to localize the secondary antibodies. For double-labeling studies, the above processes were repeated with 20% goat serum and primary c-Myc rabbit polyclonal antibodies (Santa Cruz Biotechnology) at a dilution of 1:100 overnight at 4°C, and secondary biotinylated goat anti-rabbit immunoglobulin G, followed by Texas Red Avidin D (Vector). The slides were rinsed with PBS, mounted in aqueous mounting media with DAPI and observed using fluorescein microscopy.
Double labeling with c-Myc immunohistochemistry and TUNEL
The authors performed double staining of c-Myc antibodies using c-Myc immunohistochemistry and the TUNEL (terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick-end labeling) method, as previously described (Namura et al., 1998), with minor modifications. The experimental animals were killed 24 hours after 1 hour of focal ischemia. After transcardiac perfusion, fixed sections were immunostained with c-Myc antibodies as described above. Sections were incubated with Fluorescein anti-rabbit antibodies (Vector) and mounted on glass slides (Superfrost, Fisher Scientific, Pittsburgh, PA, U.S.A.). After eliminating peroxidase activity with 3% H2O2 in PBS, the sections were stained using an in situ technique (TUNEL reaction) to detect the DNA-free 3′-OH ends (Kondo et al., 1997). Briefly, slides were placed in 1x terminal deoxynucleotidyl transferase buffer (Life Technologies, Gaithersburg, MD, U.S.A.) for 30 minutes, reacted with terminal deoxynucleotidyl transferase enzyme (Life Technologies), and biotinylated with 16-dUTP (Boehringer-Mannheim, Indianapolis, IN, U.S.A.) at 37°C for 90 minutes. Slides then were washed in 2x TB buffer (60 mmol/L NaCl, 6 mmol/L sodium citrate) and in PBS. Sections were incubated with Texas Red Avidin DCS (Vector). Slides were rinsed with PBS, mounted in aqueous mounting media, and observed using fluorescein microscopy.
Isolation of nuclear extract from brain tissues
Brain tissue from the control and ischemic mice was used to obtain nuclear protein extracts. After 60 minutes of MCAO, mice were allowed to recover for 1 hour. After the brains were cut coronally, tissue from the ipsilateral cortex, which was taken from the third to the fifth 2-mm sections from the ischemic hemisphere, and tissue from corresponding regions on the contralateral side, was separately dissected. Nuclear extracts were isolated from 250 mg of brain tissue pooled from three mice using the method of Deryckere and Gannon (1994). Briefly, the frozen tissue was broken and reduced to powder with a pestle and mortar in dry ice. All of the following steps were performed at 4°C. The thawed powder was homogenized in 5 mL solution A (0.6% Nonidet P-40, 150 mmol/L NaCl, 10 mmol/L HEPES, pH 7.9, 1 mmol/L EDTA, 0.5 mmol/L phenyl-methylsulfonyl fluoride) using a 10-mL tissue glass homogenizer (Wheaton, Millville, NJ, U.S.A.), and after 20 strokes was transferred to a 15-mL tube and centrifuged for 2 minutes at 760 g to remove any unbroken tissue. The supernatant was incubated for 5 minutes on ice and centrifuged for 10 minutes at 3000 g. The pelleted nuclei were resuspended in 4 volumes of solution B (25% glycerol, 20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 1.2 mmol/L MgCl2, 0.2 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 0.5 mmol/L phenyl-methylsulfonyl fluoride, 2 mmol/L benzamidine, plus 5 μg/mL each of the following three protease inhibitors: pepstatin, leupeptin, and aprotinin) and then incubated on ice for 20 minutes for high salt extraction. After 10 minutes of centrifugation, the supernatant containing the DNA-binding proteins then was aliquoted in small fractions and stored at −70°C. Total nuclear extract protein content was quantitated by the Bradford assay and normalized using extraction buffer.
Western blot analysis
Equal amounts (10 μg) of nuclear protein extracts were denatured at 100°C for 5 minutes in Laemmli sample buffer/5% 2-mercaptoethanol, electrophoresed on 10% polyacrylamide gels, and electroblotted onto a membrane (Novex, San Diego, CA, U.S.A.). The membrane was incubated overnight at 4°C with primary rabbit polyclonal antibodies against NF-κB p65 (1:1000) or c-Myc (1:500) (Santa Cruz Biotechnology) diluted in TBST/0.5% nonfat milk buffer (10 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 0.1% Tween-20). Then, the membrane was incubated with horseradish peroxidase conjugated secondary anti-rabbit immunoglobulin G (Boehringer Mannheim) at a 1:10000 dilution in TBST/0.5% milk buffer for 30 minutes at room temperature. The immunoreactive bands were finally visualized by the chemiluminescence detection system (ECL Plus Kit; Amersham, Buckinghamshire, England) and then the blot was exposed to Amersham x-ray film. The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, U.S.A.) and the results were quantified using Multi-Analyst software (Bio-Rad). After the first reaction, a 1:500 dilution of rabbit polyclonal antibodies against TFIID basal transcription factor (Santa Cruz Biotechnology) was used to check that the protein was loaded equally in each lane. The whole cell lysate of NIH/3T3 from a normal embryo fibroblast (Santa Cruz Biotechnology) was used as a positive antigen control of NF-κB p65 (data not shown). The proteins of amino terminal domain of human c-Myc (Santa Cruz Biotechnology) were used as positive antigen controls of c-Myc (data not shown). The membrane was incubated with antibodies against β-actin, a constitutive protein in the cytoplasm. No binding band was shown in these nuclear proteins compared with positive antigen controls, suggesting no cytoplasmic contamination in these nuclear proteins (data not shown).
Electrophoretic mobility shift assays
Double-stranded oligonucleotide probes used for protein binding in electrophoretic mobility shift assays (EMSAs) were as follows (only the upper strand is indicated): NF-κB, 5′-agttgaggggactttcccaggc-3′; Sp-1, 5′-attcgatcggggcggggcgagc-3′ (Promega, Madison, WI, U.S.A.); c-Myc, 5′-ggaagcagaccac-gtggtctgcttcc-3′ (Santa Cruz Biotechnology). Oligonucleotide probes were radiolabeled with [32P]-αATP by T4 polynucleotide kinase (USB, Cleveland, OH, U.S.A.) to produce double-stranded DNA probes. Electrophoretic mobility shift assays were performed according to the method of Singh et al. (1986). Binding reactions were conducted in a total volume of 20 μL containing equal amounts of nuclear protein (3 or 5 μg), 0.1 to 1.0 ng of DNA probe (ca. 50,000 cpm), 20 mmol/L HEPES, pH 7.9, 1.5 mmol/L MgCl2, 100 mmol/L NaCl, 1 mmol/L EDTA, 11% glycerol, 1 mmol/L dithiothreitol, 35 nmol/L phenyl-methylsulfonyl fluoride, 140 nmol/L benzamidine, and 350 ng/mL pepstatin A, leupeptin, and aprotinin for 20 minutes at 25°C. After incubation, bound and free probes were separated by 6% nondenaturing polyacrylamide gel electrophoresis and visualized by autoradiography. For competition experiments, radiolabeled DNA probes and nuclear proteins were incubated with a 100-fold molar excess of the unlabeled DNA oligonucleotide or an unrelated, unlabeled oligonucleotide. Supershift assays were performed as recommended by Santa Cruz Biotechnology by preincubating nuclear extracts with polyclonal antibodies for 60 minutes at 4°C before adding labeled probes. Polyclonal antibodies directed against p50 and p65 subunits of NF-κB were obtained from Santa Cruz Biotechnology. The hybridization signal on the x-ray film was scanned with the GS-700 imaging densitometer (Bio-Rad) and the results were quantified using Multi-Analyst software (Bio-Rad).
Statistics
The results of the densitometric analysis shown were the average of three independent experiments and expressed as mean ± SD. Statistical comparisons were performed by analysis of variance followed by Fisher's PLSD. A P value < 0.05 was considered significant.
RESULTS
Early activation of NF-κB after transient focal ischemia
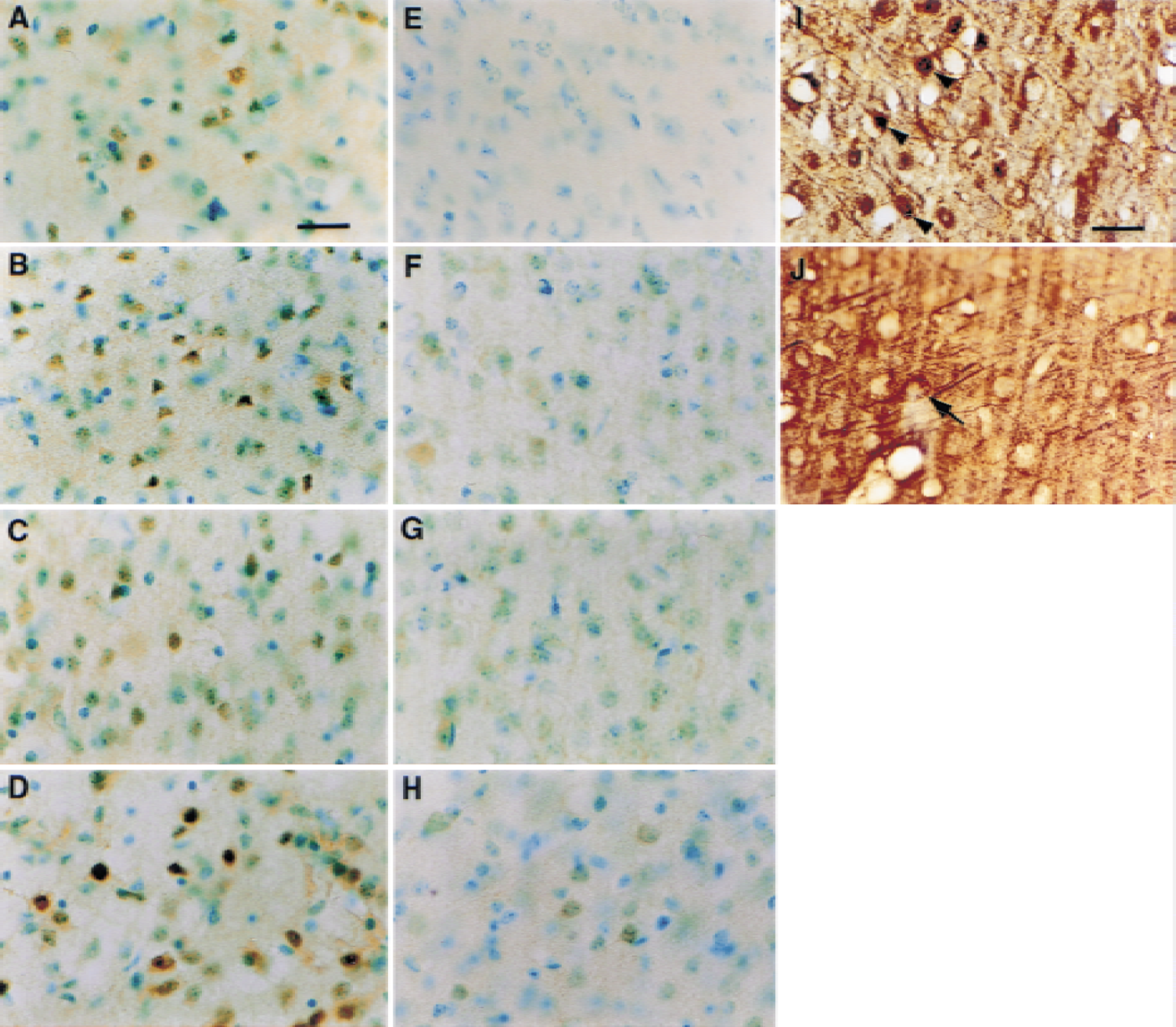
To investigate the expression of activated NF-κB after transient focal ischemia, the authors examined the temporal expression of activated NF-κB using the anti-NF-κB Ab (p65) in immunohistochemical studies. The SOD1 Tg mice and their wild-type littermates were subjected to MCAO and reperfusion. After 60 minutes of MCAO, the mice were allowed to recover for 5 minutes, and 1, 4, and 24 hours before death. The brains of the normal mice had a low level of basal constitutive NF-κB activity (Fig. 1E). Immunohistochemistry in the wild-type mice showed an acute increase in NF-κB p65 nuclear translocation in the ischemic cortex, the outer boundary of the ischemic cortex (Fig. 1A), and striatum (data not shown) as early as 5 minutes after MCAO. One hour after ischemia, the ischemic cortex and its outer boundary displayed a significantly increased level of NF-κB immunoreactivity (IR) (Fig. 1B). At 4 hours, the outer boundary of the ischemic cortex expressed induced NF-κB IR (Fig. 1C) and was maximized 24 hours after FCI (Fig. 1D). Thus, NF-κB IR in the ischemic cortex and its outer boundary was enhanced by ischemic and reperfusion injury in a time-dependent manner (Fig. 1A to 1D).
Effect of overexpression of SOD1 on NF-κB expression after FCI
To clarify the role of oxygen free radicals in the expression of NF-κB, the NF-κB IR was investigated in SOD1 Tg mice compared with their wild-type littermates after transient FCI. In the Tg mice, basal NF-κB IR remained in the ischemic cortex and its outer boundary 1, 4, and 24 hours after FCI (Fig. 1F, 1G, and 1H). Overexpression of SOD1 significantly reduced the increased level of NF-κB IR in the ischemic cortex and its outer boundary 1, 4, and 24 hours after FCI in the wild-type mice (Fig. 1B, 1C, and 1D). At the same time, the striatum within the ischemic core in the Tg mice remained intact, whereas the striatum within the ischemic core in the wild-type mice progressively lost NF-κB activity (data not shown).
Identification of cell type expressing activated NF-κB
To identify the cell types in which NF-κB was activated after FCI, the authors performed double immunohistochemical staining procedures with antibodies against the neuronal marker MAPs and p65. NF-κB p65 immunoreactivity was visualized using DAB immunohistochemistry in the first staining reaction. MAPs IR was visualized using DAB/nickel sulfate immunohistochemistry in the subsequent reaction. Results showed MAPs and p65 colocalization in the cortex on the ipsilateral side 1 hour after FCI, suggesting that NF-κB p65 was found to be up-regulated in certain neurons (Fig. 1I). Without NF-κB p65 nuclear translocation, only MAPs IR was detected in the contralateral cortex with the morphologic appearance of neurons (Fig. 1J).
Effect of overexpression of SOD1 on NF-κB DNA binding activity after FCI
Activation of NF-κB is mainly caused by increased DNA binding activity after NF-κB release from IκB. Because a significant increase in NF-κB IR was observed in the cortex on the ipsilateral side 1 hour after ischemia, the authors used EMSAs to further investigate the impact of transient focal ischemia on NF-κB DNA binding activity 1 hour after FCI. Two DNA–protein complexes could be detected in the mouse brains (Fig. 2A), which was consistent with previous findings from other groups (Schneider et al., 1999; Yu et al., 1999). The specificity of the DNA–protein complexes was verified using different unlabeled competitors. These complexes were completely abolished by competition with a 100-fold molar excess of unlabeled NF-κB oligonucleotides (Fig. 2A, lane 5), but not by competition with an unrelated oligonucleotide, Sp-1 (Fig. 2A, lane 6). In wild-type mice, ischemic and reperfusion injury increased NF-κB DNA binding activity by 154% ± 16% of control (P < 0.05) (Fig. 2A, lane 3). In the Tg mice, NF-κB DNA binding activity after ischemia was increased by 113% ± 10% of control (Fig. 2A, lane 4). These data indicate that NF-κB DNA binding activity substantially increased after ischemia in the wild-type mice and that the ischemia-enhanced DNA binding activity was reduced in the Tg mice.
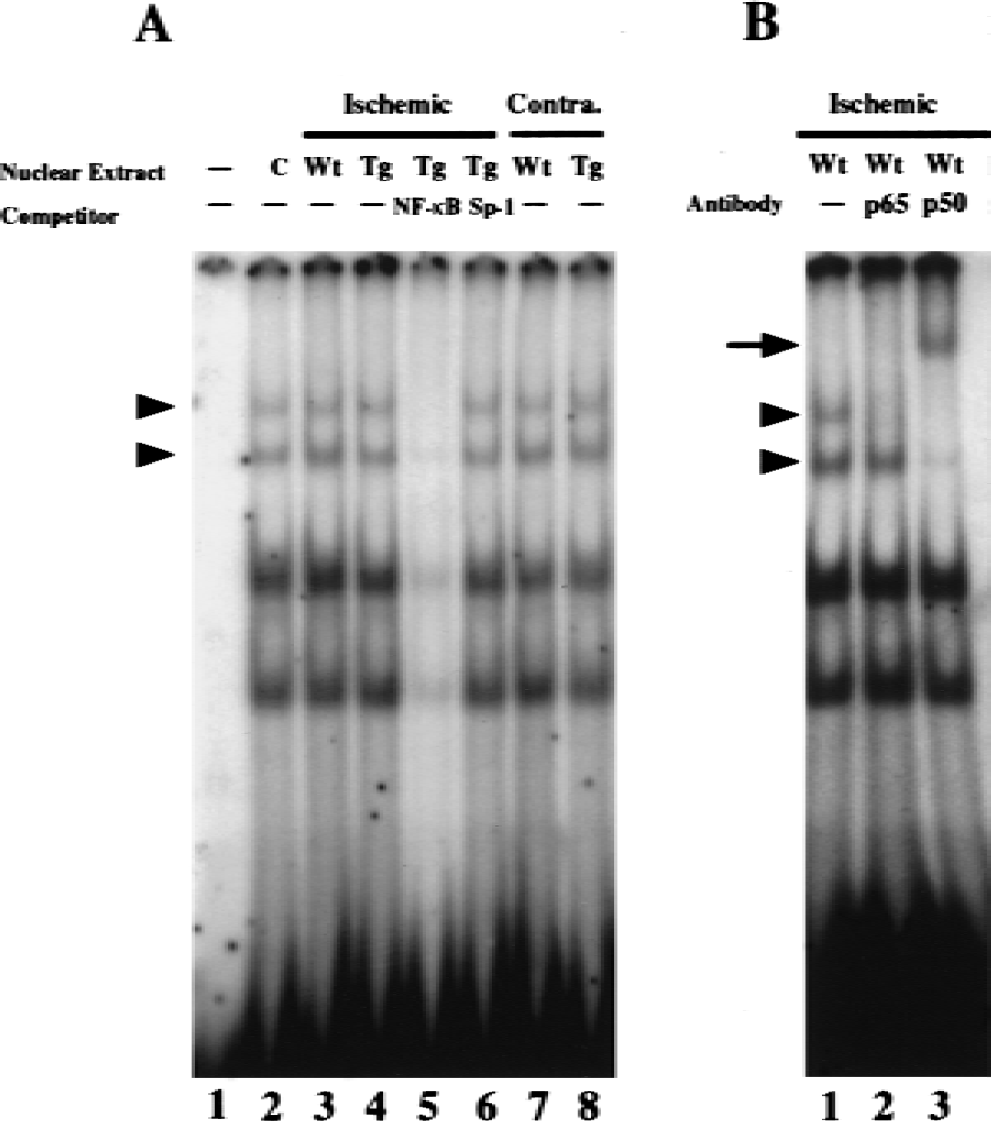
Overexpression of SOD1 decreased activated NF-κB DNA-binding activity 1 hour after focal cerebral ischemia (FCI).
Next, the composition of the two complexes was determined by performing a supershift assay with specific antibodies against various NF-κB subunits. Classic NF-κB resembles a hetero- or homodimeric protein composed of a 50-kDa subunit (p50) and a 65-kDa subunit (p65). The upper complex was abolished by antibodies against p65, suggesting that the upper complex contains p65 (Fig. 2B, lane 2). In addition, the upper complex and part of the lower complex were supershifted by antibodies against p50 (Fig. 2B, lane 3), indicating that both the upper and lower complexes contained p50. Thus, the upper complex consisted of a p50/p65 heterodimer, whereas the lower complex appears to have contained, at least partially, a p50 homodimer. The lower complex does not seem to have contained p65 because p65 antibodies had no effect on it. Although the lower complex appears to have predominantly contained p50, the possibility of involvement of other subunits that might contribute to the lower complex remains to be elucidated. Incubation of the probes with anti-p50 and anti-p65 antibodies, respectively, without nuclear extracts did not result in a supershifted complex (data not shown). Thus, supershift assays further confirmed the binding specificity of two DNA-protein complexes that contained p65 and p50 subunits. Results of increased NF-κB DNA binding activity in the ischemic cortex using EMSAs confirmed the above observation of the up-regulation of NF-κB p65 nuclear translocation in the ischemic cortex using immunohistochemistry.
Effect of overexpression of SOD1 on NF-κB p65 protein levels after FCI
Results of the EMSAs showed ischemia-enhanced NF-κB DNA binding activity. These data suggest the possibility that ischemia may activate the NF-κB protein levels in nuclear extracts. To determine whether the abundance of NF-κB protein increased in nuclear extracts, the authors performed Western blot analysis using anti-p65 polyclonal antibodies. The same nuclear extracts from the cortex that were used in the EMSAs were used and resolved by polyacrylamide gel electrophoresis. As shown in Fig. 3, the anti-p65 antibodies detected a single band with an apparent molecular weight of 65 kDa (p65) (upper panel). Compared with controls (Fig. 3, lane 1), a significant increase (624% ± 37% of control, P < 0.05) in levels of NF-κB p65 was detected 1 hour after ischemia in wild-type mice (Fig. 3, lane 2), whereas ischemia caused a smaller increase (417 ± 26% of control, P < 0.05) in extracts from the Tg mice (Fig. 3, lane 3). The reduction of increased NF-κB protein contents in Western blotting further confirms the decrease of ischemia-enhanced NF-κB in SOD1 Tg mice in immunohisto-chemistry and EMSAs, indicating that in addition to the reduction of FCI-induced NF-κB nuclear translocation and binding activity in Tg mice, a substantial decrease in transcription and/or translation of NF-κB may occur in the Tg mice after FCI. The authors observed a sixfold difference in abundance of p65 protein but only a 50% increase of DNA binding activity. It is possible that the conformation of protein may not keep totally intact through the binding reaction and gel electrophoresis. This may lead to loss of the ability of protein–DNA binding complex formation in EMSA. Moreover, NF-κB BNA binding activity may be influenced by the balance between p65 to IκB and with other subunits like p50, and posttranslational modification of subunits themselves. TFIID basal transcription factor protein was used as an internal control to show equal loading of samples (Fig. 3, lower panel). The whole cell lysate of NIH/3T3 was used as a positive antigen control of NF-κB to detect the position of p65 (data not shown).
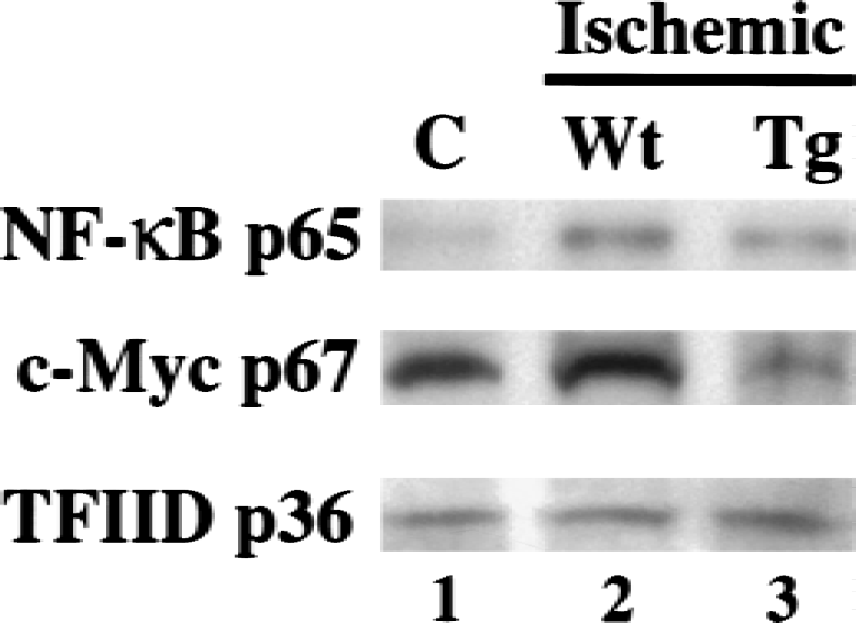
Overexpression of SOD1 decreases activated NF-κB and c-Myc protein content 1 hour after focal cerebral ischemia (FCI). Western blot analysis using nuclear extract, isolated from the control cortex (C, lane 1), wild-type ischemic cortex (Wt, lane 2), and transgenic ischemic cortex (Tg, lane 3) 1 hour after FCI. The NF-κB p65 protein was evident as a single band of molecular mass p65-kDa nuclear protein (upper panel). The c-Myc protein was evident as a single band of molecular mass p67-kDa nuclear protein (middle panel). NF-κB p65 and c-Myc protein levels were increased in the wild-type mice after FCI and were decreased in the Tg mice. TFIID basal transcription factor protein was used as an internal control (lower panel).
Functional implications of activated NF-κB after FCI
To demonstrate the functional nature of NF-κB after FCI, the authors examined the colocalization of the activated transcription factor with the expression of an established target gene, c-Myc. First, the authors examined the influence of FCI on c-Myc IR in wild-type mice. Compared with the contralateral cortex (Fig. 4A), a large increase in c-Myc IR could be detected in the outer boundary zone of the ischemic lesion 1 hour after ischemia (Fig. 4B). At 4 hours, the FCI-induced c-Myc IR could be observed in the entire ischemic cortex (Fig. 4C). At 24 hours, the ischemic cortex and striatum displayed an increase in c-Myc IR (Fig. 4D). To clarify the role of oxygen free radicals in the expression of c-Myc, the c-Myc IR was investigated in the SOD1 Tg mice compared with their wild-type littermates after transient FCI. The FCI-induced acute activation of c-Myc IR in the ischemic cortex was significantly reduced in SOD1 Tg mice 1 and 4 hours after FCI (Fig. 4E and 4F). Using a double-labeling technique, the authors demonstrated colocalization of c-Myc and the neuronal marker MAPs (data not shown), indicating that c-Myc IR was in cells that have the morphologic characteristics of neurons.
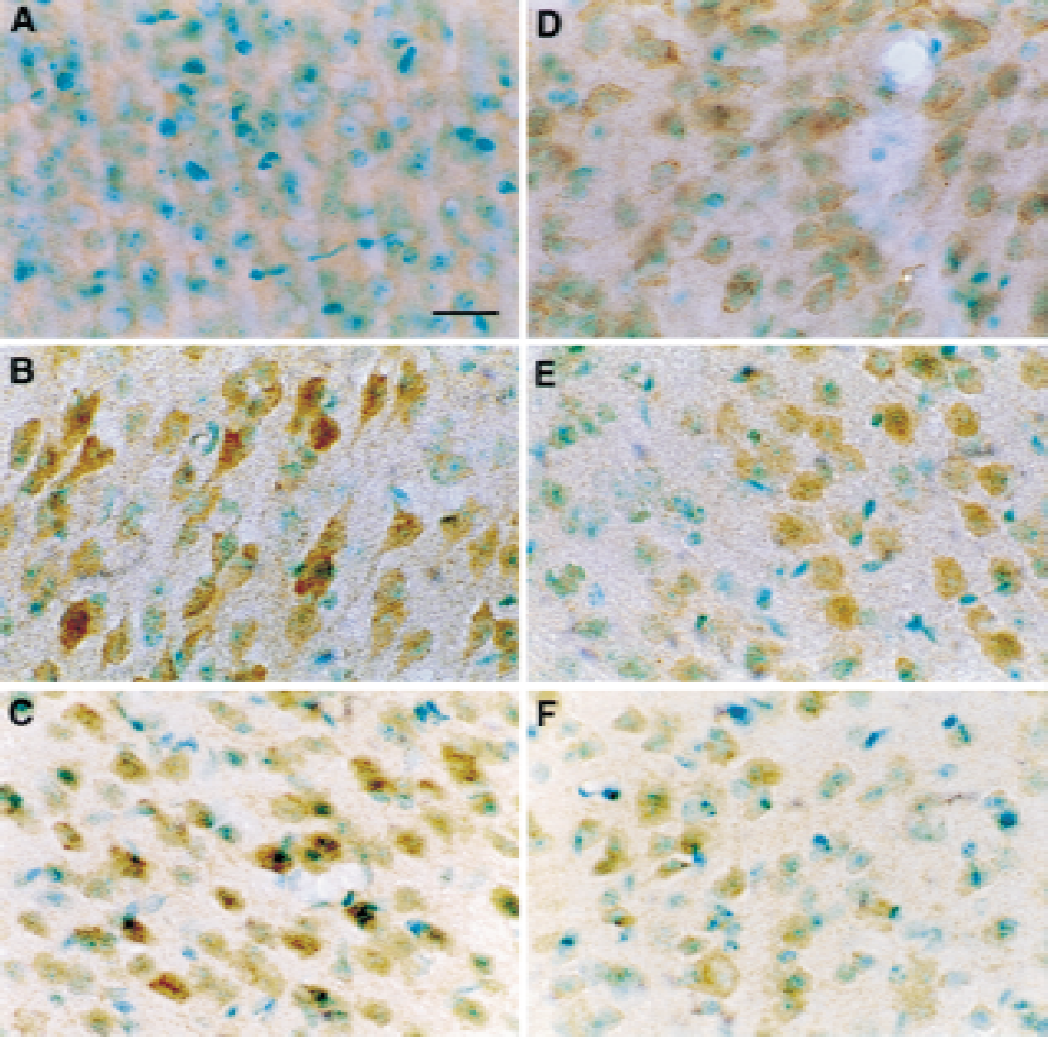
High-power views of immunohistochemical experiments to detect the temporal c-Myc expression after focal cerebral ischemia (FCI). No c-Myc immunoreactivity was detected in the contralateral cortex of wild-type mice 1 hour after FCI
To determine whether the abundance of c-Myc protein increased in nuclear extracts after FCI, the authors performed Western blot analysis, using anti-c-Myc polyclonal antibodies. As shown in Fig. 3, the anti–c-Myc antibodies detected a single band with a molecular weight of 67 kDa (middle panel). Compared with controls (Fig. 3, lane 1), an apparent increase (134% ± 17% of control) in levels of c-Myc was detected 1 hour after ischemia in wild-type mice (Fig. 3, lane 2), whereas the levels of c-Myc were decreased in extracts from the Tg mice (Fig. 3, lane 3). The reduction of increased c-Myc protein content in Western blotting further confirms the decrease of ischemia-enhanced c-Myc in SOD1 Tg mice in immunohistochemistry. TFIID basal transcription factor protein was used as an internal control as described above (Fig. 3, lower panel). The proteins of the amino terminal domain of human c-Myc were used as positive antigen controls of c-Myc (data not shown).
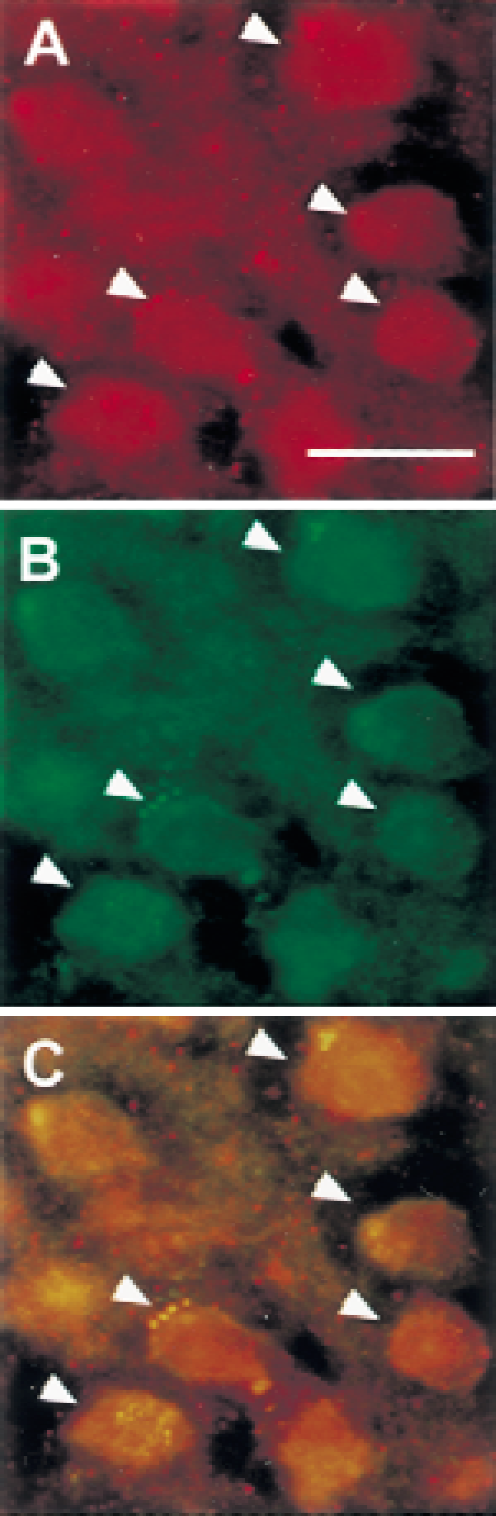
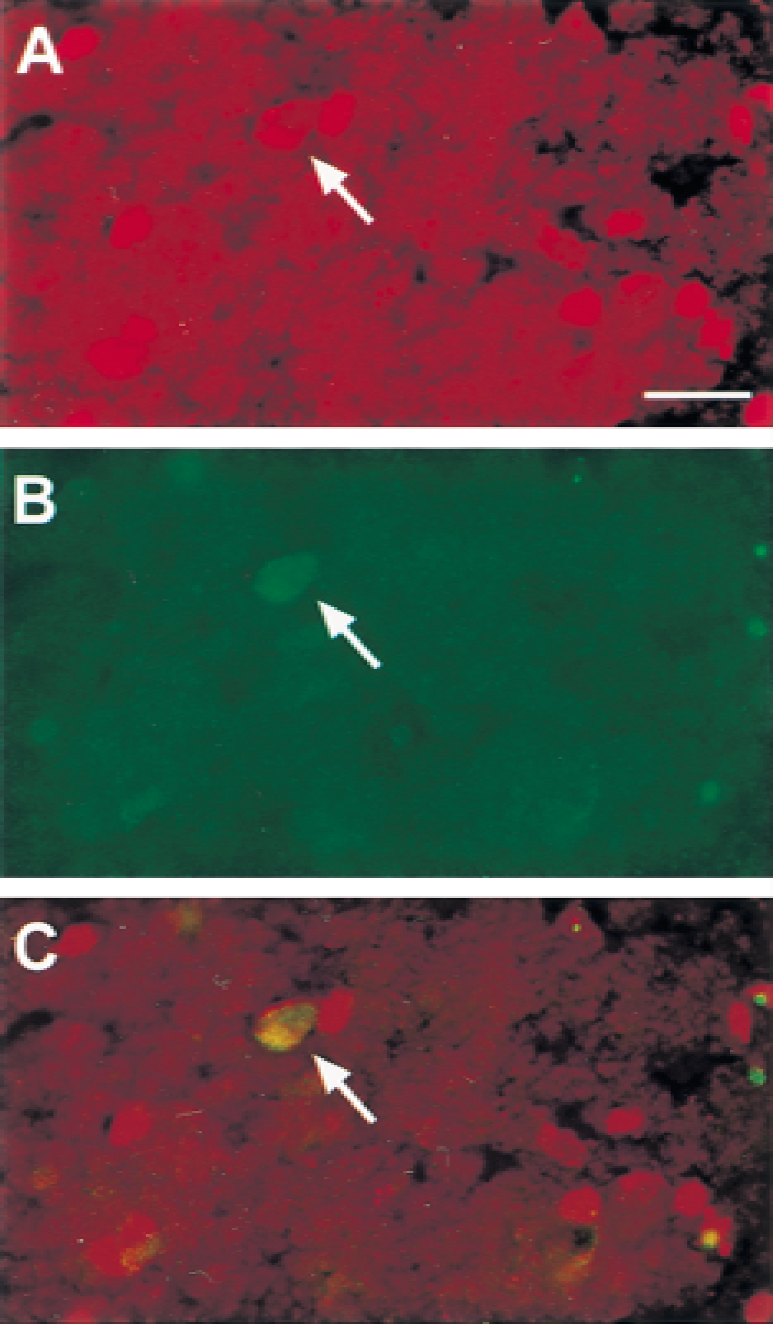
Next, the authors examined the colocalization of NF-κB and c-Myc. NF-κB activation was labeled in the first reaction and c-Myc in the subsequent step. The authors were able to detect neuronal colocalization of activated c-Myc and NF-κB in the ischemic cortex 4 hours after FCI (Fig. 5C). Thus, colocalization of activated NF-κB with c-Myc within the same cells suggests that NF-κB activation possibly participates in gene expression of c-myc after FCI.
Colocalization of activated NF-κB with the NF-κB target gene product c-Myc 4 hours after focal cerebral ischemia (FCI). Arrowheads indicate the activated transcription factors.
Double labeling with c-Myc expression and DNA fragmentation detected by TUNEL staining after FCI
In previous studies, the detection of DNA fragmentation was demonstrated by a significant amount of TUNEL-positive cells and DNA laddering 24 hours after transient focal ischemia in mice (Fujimura et al., 1999a, b ). To elucidate whether the TUNEL-positive cells in the ischemic lesion expressed c-Myc, the authors double-stained sections with c-Myc antibodies and the TUNEL technique. Twenty-four hours after FCI, a significant number of TUNEL-positive cells were detected in the ischemic striatum and some in the cortex. Some nuclei of neurons in the ischemic infarct demonstrated double immunostaining of the activated c-Myc and TUNEL (Fig. 6). Colocalization of c-Myc and TUNEL staining were observed in the same neuron, suggesting that ischemia-induced elevation of c-Myc may be involved in ischemic cellular events including neuronal cell apoptosis.
Representative photograph shows double labeling with c-Myc immunoreactivity and TUNEL staining, clarifying the anatomic relation between c-Myc expression and DNA fragmentation in the ischemic cortex 24 hours after focal cerebral ischemia (FCI).
DISCUSSION
Although NF-κB was originally studied in the immunologic and inflammatory processes, it is widely expressed in the central nervous system in both constitutive and inducible forms (Kaltschmidt et al., 1994; O'Neill and Kaltschmidt, 1997). Studies suggest a consequential role for NF-κB in a variety of injury paradigms (Devary et al., 1993; O'Neill and Kaltschmidt, 1997) and a possible deleterious role in the central nervous system. NF-κB induction has been detected in traumatic brain injury (Yang et al., 1995), spinal cord injury (Bethea et al., 1998), global forebrain ischemia (Clemens et al., 1997b), and focal ischemia (Schneider et al., 1999). In the current study, the immunohistochemical data showed an acute increase in NF-κB p65 nuclear translocation in the ischemic cortex as early as during 5 minutes of reperfusion after 1 hour of FCI (Fig. 1A). This observation is supported by studies showing that NF-κB translocation became evident in the striatum after 2 hours of FCI and 20 hours of reperfusion and that it was enhanced after 72 hours of reperfusion in the penumbral cortex (Schneider et al., 1999). The increased NF-κB protein content on Western blot analysis confirms the immunohistochemical detection of activated NF-κB after ischemia (Fig. 3). In the current study, NF-κB p65 was up-regulated in some neurons after FCI, using double-staining of p65 and MAPs (Fig. 1I). A low level of constitutive NF-κB was previously found in rat cortical and hippocampal neurons (Kaltschmidt et al., 1994). Cells that express NF-κB have the morphologic characteristics of neurons after spinal cord injury (Bethea et al., 1998). After global ischemia, NF-κB was detected in degenerating CA1 neurons (Clemens et al., 1997b). Thus, the authors' observations coincide with earlier findings that NF-κB is expressed in neurons.
The antioxidant enzyme is one of the major mechanisms by which cells counteract the deleterious effects of ROS after FCI. The authors have shown that SODs play a protective role against FCI (Chan, 1996; Kinouchi et al., 1991; Kondo et al., 1997; Murakami et al., 1998). Furthermore, there was a marked decrease in DNA fragmentation after transient FCI in Tg mice that overexpress SOD1 compared with wild-type mice (Fujimura et al., 1999b). In addition, free radicals regulate the activity of transcription factors, including AP-1 and NF-κB (Müller et al., 1997; Schulze-Osthoff et al., 1995). The current findings suggest inhibition of FCI-induced NF-κB is involved in the neuroprotective role of SOD against FCI. The authors observed a significant reduction in NF-κB IR in the Tg animals that overexpressed SOD1 compared with NF-κB IR in wild-type mice 1, 4, and especially 24 hours after FCI (Fig. 1). In addition, EMSAs showed that an increase of DNA binding activity was detected after FCI and was decreased in SOD1 Tg mice (Fig. 2A). The profile of NF-κB protein content detected on Western blots (Fig. 3) parallels that demonstrated in the EMSAs. The lesser abundance of NF-κB proteins may contribute to decreased DNA binding activity in SOD1 Tg mice. Particularly, superoxide may be involved in reperfusion brain injury after cerebral ischemia (Chan, 1994, 1996; Siesjö et al., 1989). Thus, overexpression of SOD1 suppressed ischemia-induced activation of NF-κB through a decrease in nuclear translocation, DNA binding activity, and protein levels, suggesting superoxide radicals modulate NF-kB activity after focal ischemia in a multistep process.
The authors' view is supported by other studies. In a global ischemic model, free radical scavengers inhibited ischemia-induced NF-κB activation and neuronal damage (Clemens et al., 1998), suggesting a molecular mechanism by which antioxidants can protect neurons against the deleterious effects of oxidative stress. In addition, free radical scavengers, especially effective against superoxide, attenuate quinolinic acid-induced activation of NF-κB and DNA fragmentation in vivo, suggesting that ROS contribute to this activation and that free radical scavengers partially protect striatal neurons against excitotoxin-induced apoptosis through the inhibition of NF-κB activation (Nakai et al., 1999). Furthermore, a methamphetamine-induced increase of striatal NF-κB DNA binding activity is attenuated in SOD1 Tg mice, suggesting that ROS generated by methamphetamine can activate striatal NF-κB in the monoaminergic system in the brain (Asanuma and Cadet, 1998).
Superoxide and H2O2 act as direct or indirect messengers in the activation of NF-κB in response to certain stimuli (Schmidt et al., 1995). NF-κB is activated by a prooxidant state in the cell and is therefore potentially inhibited by antioxidants (Meyer et al., 1993). Interestingly, FCI appears to induce the increase of NF-κB IR in the ischemic cortex in a time-dependent manner (Fig. 1A to 1D). Because NF-κB is a response to ROS, the increase in NF-κB could be caused by more severe oxidative stress during the longer reperfusion time. Based on the above findings, ROS may play an important role in the activation of NF-κB during reperfusion. Thus, prevention of NF-κB activation in SOD1 Tg mice contributes at least in part to the attenuation of ischemic injury.
There is increasing evidence that transcription factors of the NF-κB/Rel family are involved in apoptosis. Although an antiapoptotic role for NF-κB has been observed in certain cell types and conditions (Mattson et al., 1997; Van Antwerp et al., 1996; Wang et al., 1996), other studies have suggested that NF-κB promotes apoptosis in a variety of cell injury models (Abbadie et al., 1993; Grilli et al., 1996; Lin et al., 1995). Yu et al. (1999) found that the administration of NF-κB decoy DNA enhanced kainate-induced excitotoxic neuronal injury. In contrast, recent studies report that NF-κB may promote cell death in neurons in global ischemia (Clemens et al., 1997a) and focal ischemia (Schneider et al., 1999; Stephenson et al., 2000). The NF-κB SN 50 peptide, which blocks the NF-κB nuclear translocation, reduces quinolinic-induced (Qin et al., 1999) and kainic-induced (Nakai et al., 2000) apoptosis. NF-κB may promote proapoptotic or antiapoptotic mechanisms in the central nervous system, depending on the mechanism through which activation occurs and the circumstances in which NF-κB is activated. In addition, different combinations of NF-κB subunits determine the specificity of transcriptional activation (Perkins et al., 1992). Whether NF-κB is neuroprotective (Mattson et al., 1997; Van Antwerp et al., 1996; Wang et al., 1996) might also depend on which NF-κB subunits are activated—for example, c-Rel can induce apoptosis and is strongly activated in apoptotic neurons (Abbadie et al., 1993). The authors' supershift data showed that p50 homodimers and p65/p50 heterodimers are elevated after FCI (Fig. 2B). The combination of the different subunits may have an impact on the role of NF-κB in the pathogenesis of FCI. Because the exact mechanism by which NF-κB contributes to neuronal death is unclear, it is important to clarify downstream genes that are driven by NF-κB. Among the genes induced at apoptosis, potential target genes for NF-κB include c-myc. For example, in the platelet-derived growth factor signaling pathway, NF-κB transmits a signal required for the induction of c-Myc expression and an antiapoptotic signal that counterbalances c-Myc cytotoxicity (Romashkova and Makarov, 1999), conceivably by promoting the expression of protective genes (Lee et al., 1999).
In the current study, the authors provide the first evidence that c-Myc IR is activated in transient FCI in mice (Fig. 4B to 4D). In permanent focal ischemia, mRNA expression of c-myc is markedly expressed in the cortex and hippocampus after MCAO in the rat (Nakagomi et al., 1996). These data support the authors' immunohistochemical studies. In addition, SOD1 overexpression reduced the increase of c-Myc IR after FCI (Fig. 4E to 4F). Like NF-κB, B-Myc was up-regulated in cortical neurons (data not shown). Furthermore, Western blot data showed c-Myc induction after FCI and down-regulation of c-Myc induction by SOD1 overexpression, suggesting NF-κB and c-Myc are coupled (Fig. 3). In addition, colocalization of NF-κB and the gene product, c-Myc, was detected after FCI, suggesting the functional implication of NF-κB activation after FCI (Fig. 5). The correlation between these two genes may occur at different levels, such as transcriptional regulation and posttranslational interactions. Because two functional NF-κB sites were identified within the c-myc promoter, the authors speculated that c-myc could be a downstream target gene of NF-κB by the colocalization of NF-κB and c-myc as their first attempt. Further investigations are necessary to clarify whether NF-κB acts as a transcriptional activator of c-myc. The correlation between these two genes at other levels should not be ruled out. Transient global ischemia induced IR of c-Myc and p53, potent apoptotic inducers, in susceptible CA1 neurons (McGahan et al., 1998). Apoptotic cells were observed in this region after forebrain ischemia (Chan et al., 1998; Petito et al., 1997), suggesting c-Myc may play a role in the processes of ischemic neuronal apoptosis. Consistent with this concept is the finding that some TUNEL-positive cells were also c-Myc-positive (Fig. 6). Although colocalization of c-Myc with a cell death marker does not prove that c-Myc is involved in ischemia-induced apoptosis, it does suggest an involvement in this process.
The authors' observations are supported by recent studies showing that NF-κB appears to promote quinolic acid-induced apoptosis in striatal neurons through up-regulation of c-Myc and p53 (Qin et al., 1999), and that free radical scavengers block quinolic acid-induced NF-κB, c-Myc, and p53 expression (Nakai et al., 1999). Whereas only a small fraction of TUNEL-positive cells are c-Myc-positive, other mechanisms by which FCI induces apoptosis appear to exist. For example, the rapid decrease of the DNA repair enzyme, APE/Ref-1, has a role in apoptosis after transient FCI (Fujimura et al., 1999b). The authors have demonstrated that FCI injury, which activates NF-κB, can also activate c-Myc within the same cell, indicating that ischemia-induced NF-κB may transcriptionally activate c-myc, which may contribute to neuronal cell death.
Other than this c-Myc pathway, cellular nitric oxide (NO) may provide another control mechanism for modulating SOD1 neuroprotection. Superoxide rapidly reacts with NO to form peroxynitrite, which is a strong oxidant and could form a species with the reactivity of hydroxyl radical during decomposition (Beckman et al., 1990). Thus, a high level of SOD1 activity may reduce the amount of superoxide anion, leading to an increase in the NO level by prolonging the half-life of NO itself. In addition, a decrease in superoxide anion may result in a smaller amount of peroxynitrite and hydroxyl radical, which can diminish endothelial injury and increase NO production. The protective effect in SOD1 animals could result from the reduction of peroxynitrite compared with wild-type mice (Kamii et al., 1996). On the other hand, it has been demonstrated that NO suppresses NF-κB activation by its induction and stabilization of IκB (Matthews et al., 1996; Peng et al., 1995). Therefore, it is possible that SOD1 overexpression leads to neuroprotection by increasing NO (or increasing peroxynitrite), stabilizing IκB, and preventing translocation to the nucleus.
In conclusion, an increase in NF-κB activation occurs after FCI, but to the authors' knowledge, the current study is the first to report that ROS contributed to the ischemia-induced early activation of NF-κB and that c-Myc was activated after transient FCI. The current results imply that overexpression of SOD1 decreases FCI-induced free radicals and that this may prevent the activation of NF-κB, thereby blocking the expression of downstream deleterious genes like c-myc, a potent modulator of apoptosis. The current results provide insights about signaling pathways that may mediate SOD1 neuronal protection against ischemia and a basis for therapeutic interventions.
Footnotes
Acknowledgments:
The authors thank G.W. Kim, L.F. Reola, B.E. Calagui, and B. Houle for their expert technical assistance and C. Christensen for editorial assistance.