
Abstract
Nuclear factor-κB (NF-κB) has a central role in coordinating the expression of a wide variety of genes that control cerebral ischemia. Although there has been intense research on NF-κB, its mechanisms in the ischemic brain have not been clearly elucidated. We investigated the temporal profile of NF-κB-related genes using a complementary DNA array method in wild-type mice and human copper/zinc-superoxide dismutase transgenic (SOD1 Tg) mice that had low-level reactive oxygen species (ROS) by scavenging superoxide. Our DNA array showed that IκB kinase (IKK) complex (IKKα, β, and γ) mRNA in the wild-type mice was decreased as early as 1 h after reperfusion, after 30 mins of transient focal cerebral ischemia (tFCI). In contrast, tFCI in the SOD1 Tg mice caused an increase in the IKK complex. The IKK complex protein levels were also drastically decreased at 1 h in the wild-type mice, but did not change in the SOD1 Tg mice throughout the 7 days. Electrophoretic mobility shift assay revealed activation of NF-κB DNA binding after tFCI in the wild-type mice. Nuclear factor-κB activation occurred at the same time, as did the phosphorylation and degradation of the inhibitory protein κBα. However, SOD1 prevented NF-κB activation, and phosphorylation and degradation of IκBα after tFCI. Superoxide production and ubiquitinated protein in the SOD1 Tg mice were also lower than in the wild-type mice after tFCI. These results suggest that ROS are implicated in transient downregulation of IKKα, β, and γ in cerebral ischemia.
Introduction
The nuclear factor-κB (NF-κB) family of transcription factors plays multiple roles in the inducible expression of genes involved in diverse biological processes, including development, immune/inflammatory responses, stress responses, and carcinogenesis. Nuclear factor-κB is activated in response to physical and chemical stress, oxidative stress, proinflammatory cytokines, and bacterial lipopoly***saccharide (Rothwarf, 1999 and Karin, 1999; Courtois et al, 2001; Karin and Lin, 2002; Li and Verma, 2002). In resting cells, most of NF-κB is sequestered in the cytoplasm in complexes with IkB inhibitory proteins. Once freed from IκB, NF-κB translocates into the nucleus and activates the transcription of cognate genes (Chen et al, 1996; Whiteside and Israël, 1997). Recent findings show that the IκB kinase (IKK) complex, composed of at least three subunits, catalytic IKKα and IKKβ, and regulatory IKKγ, critically phosphorylates IκB proteins (Scheidereit, 1998; Karin, 1999; Courtois et al, 2001). Phosphorylation of IκBs leads to protein-ubiquitination, which is followed by proteasome-mediated degradation.
The central nervous system is particularly vulnerable to oxidative stress because of a high rate of oxidative metabolism, which results in high rates of strong oxidant formation. In addition, the central nervous system contains an abundance of polyunsaturated fatty acids that are susceptible to lipid peroxidation. The cerebral ischemia brain is characterized by a severe reduction in blood flow. After cerebral ischemia, reperfusion can cause additional brain injury from excessive production of reactive oxygen species (ROS). Oxidative stress is a major source of injury from cerebral ischemia and reperfusion (Bowler et al, 2002). We have previously shown that less oxidative stress reduced ischemic damage in human copper/zinc-superoxide dismutase (SOD1) transgenic (Tg) mice with a three-fold increase in human SOD1, which is associated with low levels of ROS, superoxide in particular. Increased endogenous SOD activity also resulted in higher antioxidant levels (reduced glutathione and ascorbate levels) in the damaged brain, and significantly reduced the infarct size and brain edema in SOD1 Tg mice compared with wild-type mice (Kinouchi et al, 1991). Our results indicate that superoxide radicals play an important role in the pathogenesis in the brain after transient focal cerebral ischemia (tFCI).
Schneider et al (1999) reported a cell death-promoting role for NF-κB in focal ischemia. DNA binding of NF-κB subunits (p65 and p50) enhanced after 2 h of FCI and 20 h of reperfusion. Ischemic damage was significantly reduced in p50 knockout mice. There are reports of activation of NF-κB after tFCI and global cerebral ischemia (Salminen et al, 1995; Clemens et al, 1997; Gabriel et al, 1999; Stephenson et al, 2000; Huang et al, 2001) and of the neuroprotective role of antioxidants (Carroll et al, 1998; Clemens, 2000). However, Irving et al (2000) showed a decrease in NF-κB 6 h after an initial increase at 3 h in permanent FCI. Mattson and colleagues (Barger and Mattson, 1996; Yu et al, 1999, 2000) also have shown the neuroprotective role of the p50 subunit in several diseases such as Huntington's and Alzheimer and after excitotoxic injury.
The NF-κB transduction cascade is activated by stimuli such as oxidative stress and cytokines, leading to the phosphorylation of IκBα on Ser32 and Ser36 by the IKK complex. IκB kinase-mediated phosphorylation triggers the ubiquitination of IκBα (Chen et al, 1995).
The IKK complex is a converging point of NF-κB activation by a large number of stimuli. Although one of the most specific steps in NF-κB signaling is the activation of the IKK complex, how this complex is involved with this transcription factor in transient cerebral ischemia is not quite understood. IKKκ and IKKβ contain a redox-sensitive cysteine residue (cys179) in this domain, which is sensitive to oxidative modification by ROS (Kapahi et al, 2000; Rossi et al, 2000; Chen and Shi, 2002). How ROS feed into the IKK pathway in cerebral ischemia is largely unknown. Using complementary DNA (cDNA) array technology, we provide the first evidence of transient decreases in IKK complex mRNA after tFCI. This cDNA array approach was validated by Western blotting. The present study was designed to clarify the role of ROS in the IKK complex after tFCI. Our results suggest that ROS are implicated in transient downregulation of the IKK complex, the upstream components of NF-κB signaling.
Materials and methods
Transient Focal Cerebral Ischemia
Experiments were performed in accordance with National Institutes of Health guidelines and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Transgenic mice of the TgHS/SF-218 strain, which carries the SOD1 gene with a CD-1 background, were derived from the founder stock (Epstein et al, 1987). There were no observable phenotypic differences between the Tg mice and their wild-type normal littermates. Transgenic mice (3-month-old males, 35 to 40 g), with a three-fold overexpression in SOD1 activity in brain cells (Epstein et al, 1987), and wild-type mice were subjected to tFCI and reperfusion in a randomized, blind manner. Transient focal cerebral ischemia was induced by intraluminal middle cerebral artery (MCA) occlusion with a nylon monofilament suture as described previously (Yang et al, 1994). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37°C with a homeothermic blanket. The left femoral artery was cannulated for measurement of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation, 10 mins after occlusion, and 10 mins after reperfusion. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics Ltd, Essex, UK). The left common carotid artery was exposed and the left external carotid artery and its branches were electrocoagulated. An 11.0-mm 5 to 0 surgical monofilament nylon suture, blunted at the tip, was introduced into the left internal carotid artery through the external carotid artery stump. After 30 mins of proximal MCA occlusion, blood flow was restored by removing the suture. Control normal wild-type mice did not undergo surgery. After 30 mins of ischemia, cerebral blood flow was restored by removal of the nylon suture. Physiologic parameters were monitored throughout the studies and values were the same as previously reported (Fujimura et al, 1999).
cDNA Array Study
The animals were deeply anesthetized with isoflurane 1, 6, and 24 h and 7 days after cerebral blood flow restoration (n = 4). Sham-operated animals underwent exposure of carotid arteries without nylon suture insertion (n = 4). After decapitation, the brains were quickly removed and the perfused MCA territory was obtained. It was immediately frozen in powdered dry ice and kept at -80°C until use. Extraction of total RNA was performed using TRIzol reagent (15596-026; Invitrogen, Carlsbad, CA, USA) in accordance with the instructions of the manufacturer. Single-strand cDNA was synthesized by reverse transcription of the RNA (3 μg) at 42°C for 90 mins with use of biotin-16-dUTP (1093070; Roche, Mannheim, Germany), M-MLV reverse transcriptase (M1701; Promega, Madison, WI, USA), and primer mix from the NF-κB Signaling Pathway Genearray kit (MM-016N; SuperArray, Bethesda, MD, USA). Labeling, hybridization, and washing of the membrane were performed according to the manufacturer's instructions. Subsequently, the membranes were treated with CDP-star (MS050R; Applied Biosystems, Bedford, MA, USA) and exposed on X-ray film. The film was scanned and densitometries were performed with the use of Multi-Analyst software (ST32151N; Bio-Rad, Hercules, CA, USA). The spot density of the internal control housekeeping genes (i.e., β-actin and RPL13A) on the membrane was also determined, and the ratio of each gene to the housekeeping gene was calculated. This ratio was obtained from each animal, and the data from the ischemic brains at each time point were compared with those from the sham-operated brains.
Real-Time Polymerase Chain Reaction Conditions
All real-time polymerase chain reactions (PCRs) were peeformed using the M × 3000 PCR System (Stratagene, La Jolla, CA, USA). All PCR amplifications were performed using the recommended buffer supplied by the manufacturer. Polymerase chain reaction mixture consisted of 2 μL of each template (n = 4), 5 pmol of each primer, and QuantiTect SYBR Green (204143, Qiagen, Valencia, CA, USA) including ROX as an internal control. Triplicate reactions were performed for each template amount. Polymerase chain reaction cycling conditions were as follows: initial denaturation at 95°C for 2 mins, followed by 40 cycles at 95°C for 15 secs, 1 min annealing at 60°C, and a 30-sec extension at 72°C. Data analysis was performed using M × 3000 software. CT's were determined using the signal/noise ratio set to standard deviation above background-subtracted mean fluorescence values (dRn).
Western Blotting
The animals were decapitated 1, 6, and 24 h, and 21 days after reperfusion under deep anesthesia with isoflurane (n = 4). Samples from the sham-operated animals were also obtained (n = 4). Samples were obtained from the MCA territory brain tissue on the ischemic sides, including the striatum and penumbra, and were quickly frozen in powdered dry ice and kept at -80°C until use. For protein extraction, the tissue was homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ, USA) in seven volumes of cold suspension buffer (20 mmol/L HEPES potassium hydroxide (pH 7.5), 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L ethyleneglycotetraacetic acid, and 0.7% protease inhibitor cocktail (P8340; Sigma-Aldrich Corp., St Louis, MO, USA)). The cytoskeletal fraction for ubiquitinated protein was prepared from ischemic brain using ProteoExtract (539790; Calbiochem, La Jolla, CA, USA). The homogenate was centrifuged at 10,000 g for 15 mins at 4°C, and the supernatant was used for this study. Assays to determine the protein concentration were performed by comparison with a known concentration of bovine serum albumin using a kit (23227; Pierce, Rockford, IL, USA). A lysate equivalent to 10 μg of protein from each fraction was run on a sodium dodecyl sulfate-polyacrylamide gel for 120 mins at 100 V, together with a size marker (RPN800; Amersham, Piscataway, NJ, USA). The protein on the gel was subsequently transferred to a polyvinylidene fluoride transfer membrane (LC2002; Invitrogen) in a buffer containing methanol, glycine, Tris base, and sodium dodecyl sulfate, after which the membrane was placed in 5% powdered milk in Tween phosphate-buffered saline (phosphate-buffered saline with 0.1% Tween 20 (Bio-Rad)) for 1 h to block nonspecific binding. It was then incubated with primary antibodies for 12 h at 4°C. The primary antibodies used were 1:500 dilution of mouse monoclonal antibodies against IKKα, β, and γ antibody (05 to 536, 05 to 535, and 05 to 631, respectively; Upstate, Lake Placid, NY, USA), 1:200 dilution of mouse polyclonal antibody against phospho-IκBα at serine 32/36 (9246; Cell Signaling Technology, Beverly, MA, USA), 1:400 dilution of rabbit polyclonal antibody against IκBa (9242; Cell Signaling Technology), 1:1,000 dilution of anti-ubiquitin monoclonal antibody (3936; Cell Signaling Technology), or 1:10,000 dilution of anti-β-actin monoclonal antibody (A5411; Sigma). After washing, the membrane was incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Amersham International, Buckinghamshire, UK) or horseradish peroxidase-conjugated anti-rabbit immunoglobulin G at 1:5,000 dilution for 60 mins. The signal was then detected with a chemiluminescent kit (Pierce). Multi-Analyst software (Bio-Rad) was used for data analysis.
Electrophoretic Mobility Shift Assays
The animals were decapitated 1, 6, and 24 h, and 21 days after reperfusion under deep anesthesia with isoflurane (n = 4). Nuclear extracts for electrophoretic mobility shift assay were prepared from the ischemic brain using ProteoExtract (539790; Calbiochem). Nuclear factor-κB binding consensus sequence (5′-AGT TGA GGG GAC TTT CCC AGG C −3′; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was radiolabeled with [32P]-γ adenosine triphosphate by T4 polynucleotide kinase (Invitrogen) to produce double-stranded DNA probes. For binding reaction, 10 μg of nuclear extract was incubated with poly (dI***dC) and the 32P-labeled double-strand oligonucleotide (1 ng, ≥1 × 105 cpm). The reaction products were fractionated on a nondenaturing 6% polyacrylamide gel, which was then dried and subjected to autoradiography. For competition assays, the excess oligonucleotide (100-fold molar excess) competitor was preincubated with nuclear extracts for 20 mins at room temperature. A mutant NF-κB oligonucleotide used for the competition assay was as follows: 5′-AGT TGA GG
In Situ Detection of Superoxide Anion Production
The early production of superoxide anions (O−2) in cerebral ischemia was investigated using hydroethidine (HEt) via a previously described method (Saito et al, 2003). Hydroethidine is diffusible into the central nervous system parenchyma after an intravenous injection and is selectively oxidized to ethidium by O−2, but not by other ROS such as hydrogen peroxide, hydroxyl radicals, or peroxynitrite. Hydroethidine solution (200 L; 1 mg/mg in phosphate-buffered saline) was administered intravenously 15 mins before ischemia induction as described. In the brains of the animals intravenously injected with HEt, fluorescence was assessed microscopically at excitation 355 nm and emission >415 nm for HEt detection or at excitation 510 to 550 nm and emission >580 nm for ethidium detection. Animals were killed 1 h after tFCI by transcardial perfusion as described (n = 3 each). After fixation with 4% paraformaldehyde for 12 h, the brains were sectioned at 50 m on a vibratome. Subsequently, the slides were covered with Vectashield mounting medium with 4′,6 diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA). These sections were observed with a microscope under fluorescent light.
Quantification and Statistical Analysis
To evaluate the results of the Western blot and oxidative protein damage studies, the film was scanned with an imaging densitometer (GS-700; Bio-Rad) and the optical density was quantified using Multi-Analyst software (Bio-Rad) as previously reported (Noshita et al, 2001). The data are expressed as mean±s.d. Comparisons among multiple groups were performed by one-way ANOVA with appropriate Bonferroni or Dunnet's tests (GraphPad Prism; Oberlin, San Diego, CA, USA). P-values less than 0.05 were considered statistically significant.
Results
Decreases in IKK Complex (IKKα, β, and γ) mRNA as Early as 1 h After tFCI in the Wild-Type Mice were Prevented in the SOD1 Tg Mice
To investigate NF-κB-related genes in the brain that were differentially regulated by tFCI, we prepared total RNA from wild-type mice at 1, 6, and 24 h, and 7 days. Nuclear factor-κB Signaling Pathway Genearray was used to assess altered temporal gene expression profiles. Expression of IKK complex (IKKα, β, and γ) mRNA showed decreases at 1 h with a statistical significance after tFCI, but a gradual recovery to the baseline by 7 days (Figure 1A). To investigate the role of ROS in the IKK complex after tFCI, we used SOD1 Tg mice, which had a three-fold increase in human SOD1 and with concomitant low levels of ROS such as superoxide (Chan, 1996). In contrast to the wild-type mice, tFCI in the SOD1 Tg mice caused an increase in IKKα, β, and γ mRNA from 1 h, but returned to the basal level at 7 days (Figure 1B). The IKK complex is a converging point of NF-κB activation by a large number of stimuli. The role of the IKK complex is important for initiating IκB phosphorylation and subsequent NF-κB activation.
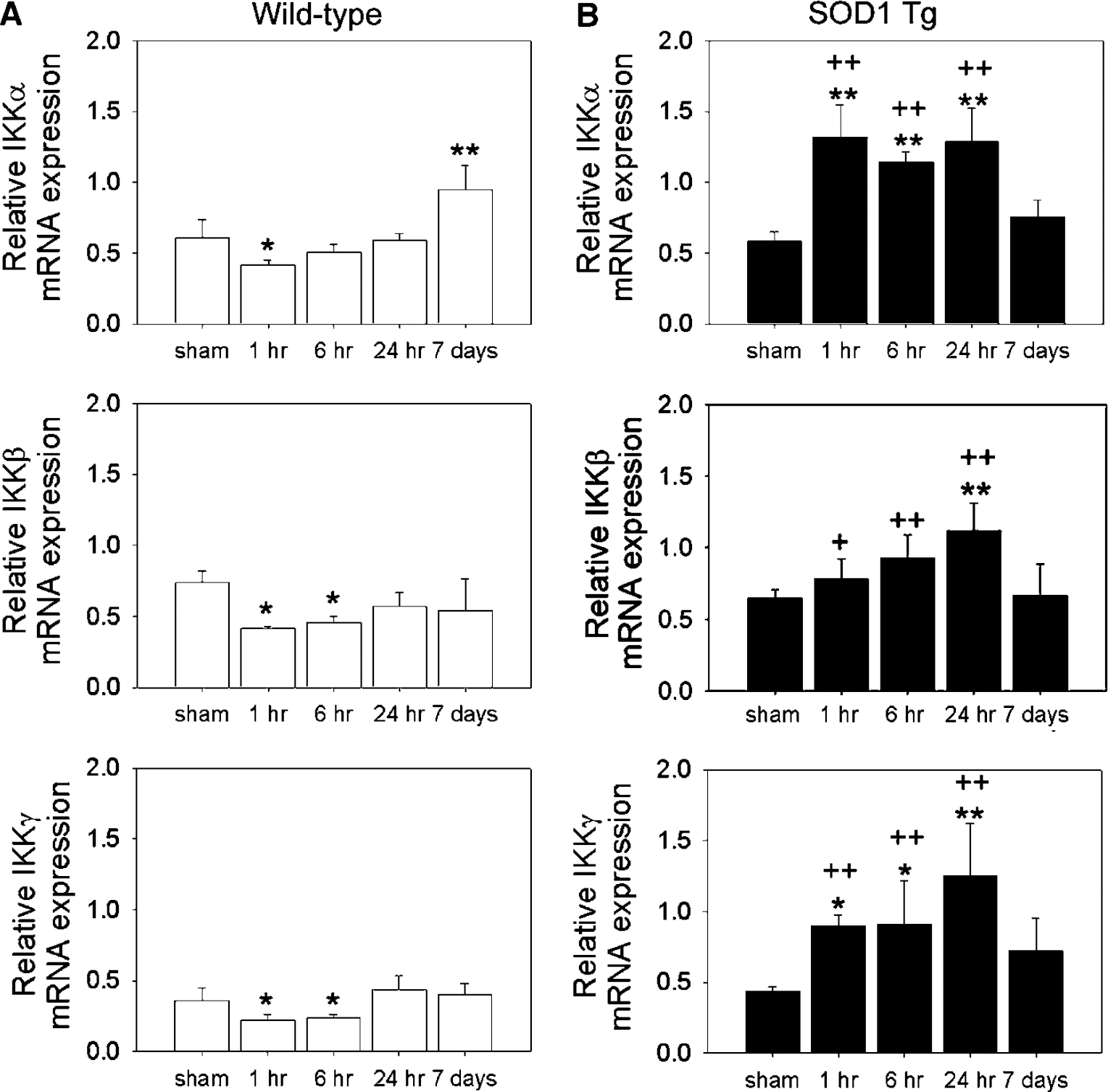
Time course of IKKα, β, and γ gene expression after 30 mins of transient focal cerebral ischemia (tFCI) in wild-type and SOD1 Tg mice. Time points include sham and 1, 6, and 24 h, and 7 days after tFCI. (
Decreases in IKK Complex Proteins After tFCI in the Wild-Type Mice were Attenuated in the SOD1 Tg Mice
To examine the protein expression of the IKK complex in the wild-type and SOD1 Tg mice, IKKα, β, and γ proteins were assessed by Western blotting, which revealed that IKKα, β, and γ immunoreactivity was evident as a single band of molecular mass of 85, 87, and 48 kDa, respectively (Figure 2). The IKKα, β, and γ protein levels were also drastically decreased at 1 h, but returned to the basal level at 7 days in the wild-type mice (Figure 2A). However, in the SOD1 Tg mice, the IKKα, β, and γ protein levels did not change throughout the 7 days after tFCI (Figure 2B). In contrast to the wild-type mice, IKKα, β, and γ protein levels were not affected by tFCI in the SOD1 Tg animals. IκB kinase decreases obtained by the cDNA microarray approach were validated by Western blotting, indicating that IKKα, β, and γ were transcriptionally regulated during reperfusion after tFCI in the wild-type mice.
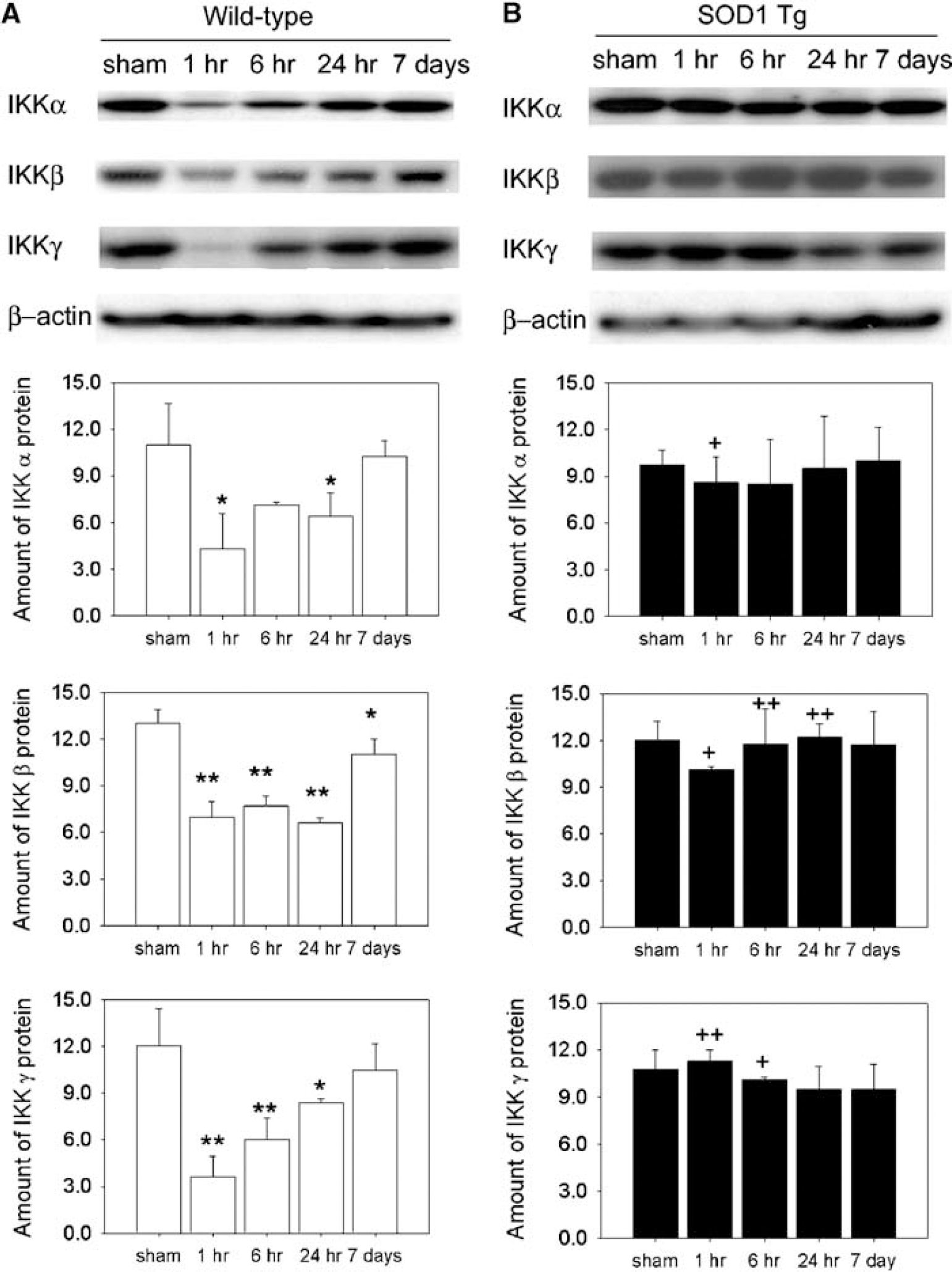
Time course of IKKα, β, and γ protein expression after 30 mins of transient focal cerebral ischemia (tFCI) in wild-type and SOD1 Tg mice. Time points include sham and 1, 6, and 24 h, and 7 days after tFCI. (
NF-κB Activation After tFCI in the Wild-Type Mice was Attenuated in the SOD1 Tg Mice
How the upstream component of the NF-κB pathway, the IKK complex, relates to NF-κB binding activity in cerebral ischemia is still unknown. Therefore, NF-κB binding activity was measured by electrophoretic mobility shift assay in nuclear extracts prepared from ischemic brains after tFCI (Figure 3A). Ischemia and reperfusion injury in the wild-type mice increased NF-κB DNA binding activity by 143%, 127%, and 110% of the control at 1, 6, and 24 h, respectively (Figure 3A). However, in the SOD1 Tg mice, NF-κB activation (Figure 3B) was prevented after tFCI. The specificity of bands depicts the binding of NF-κB in the presence of 100-fold molar excess of NF-κB unlabeled competitor (NF-κB) and anti-p65. Free probes appeared in the bottom of the gel (Figure 3C). Because phosphorylation and degradation of the inhibitory protein IκBα are essential steps for releasing NF-κB and its subsequent activation, phosphorylation and degradation of IκBα were measured by Western blotting.
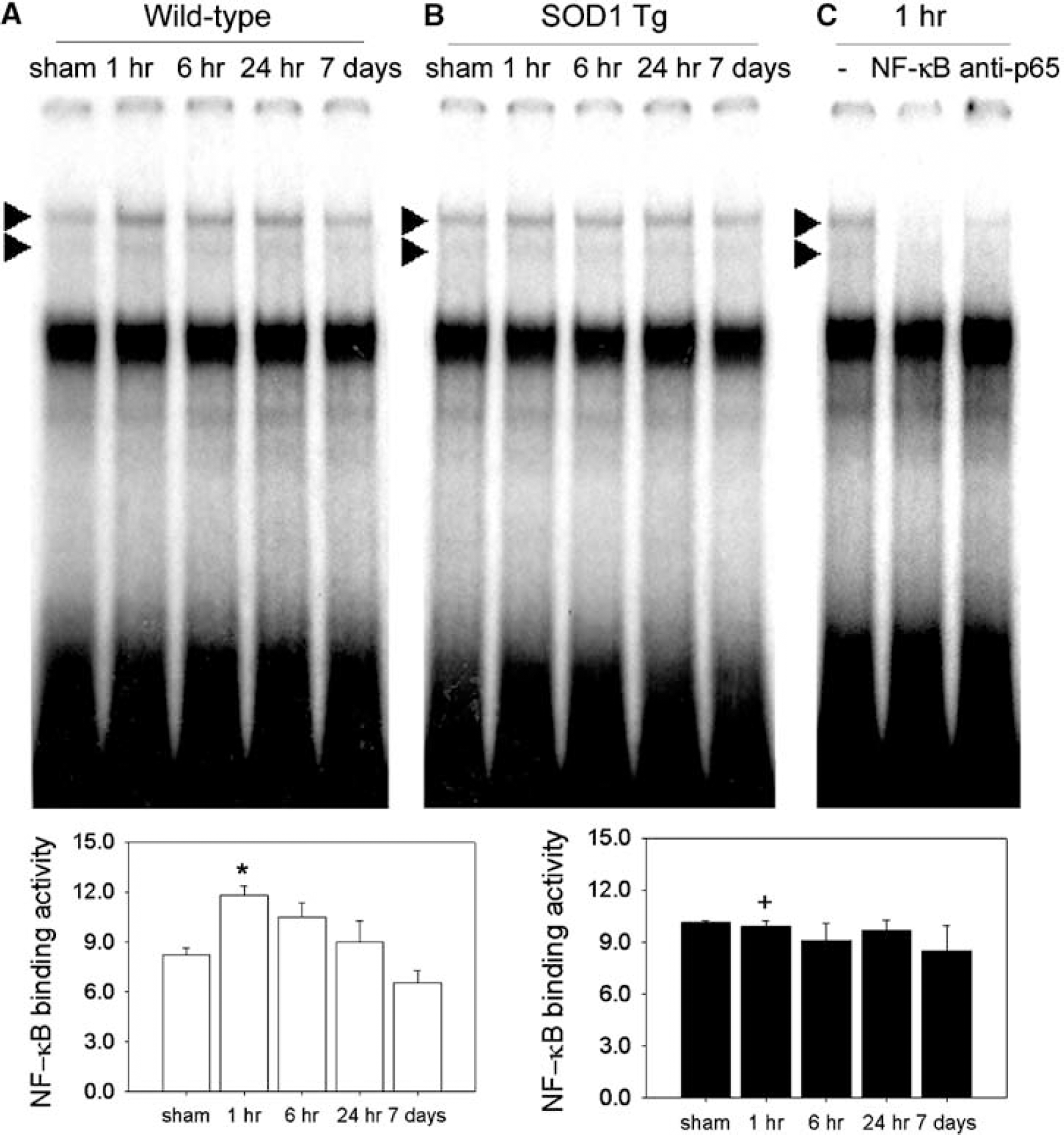
Changes in nuclear factor-κB (NF-κB) activation after 30 mins of transient focal cerebral ischemia (tFCI) in the wild-type and SOD1 Tg mice. (
SOD1 Reduced the Phosphorylation and Degradation of IκBα After tFCI
The phosphorylated form of IκBα (Ser32/36) was hardly detectable in the sham-operated mice. When the brain was subjected to tFCI, phosphorylation of IκBα started to increase at 1 h, peaked at 6 h, and returned to the basal level 7 days after tFCI (Figure 4A). IκBα was degraded, but then recovered rapidly after 6 h because the IκBα gene is one of the NF-κB-activated targets. The amount of IκBα mRNA also decreased slightly at 1 h, but increased by 24 h (Figure 4A). Phosphorylation of Ser32 and Ser36 of IκBα in the wild-type mice after tFCI revealed that the IKK complex is involved in activation of NF-κB DNA binding activity after tFCI. Phosphorylated IκBα was then ubiquitinated; this ubiquitination targeted it for degradation by the 26S proteasome. Nuclear factor-κB activation after tFCI occurred with phosphorylation and degradation of the inhibitory protein IκBα in the wild-type mice. In the SOD1 Tg mice, phosphorylation (Ser32/36) and degradation of IκBα were prevented after tFCI (Figure 4B). The amount of IκBα mRNA also decreased slightly at 1 h, but increased by 24 h (Figure 4B). However, the increase in IκBα mRNA in the SOD1 Tg mice after tFCI was less than in the wild-type mice at 24 h. These results indicate that decreases in the IKK complex accompanied IκBα degradation and subsequent NF-κB activation in the wild-type mice. As shown in Figures 3B and Figure 4B, SOD1 attenuated the IKK complex decrease by preventing IκBα degradation and NF-κB activation.
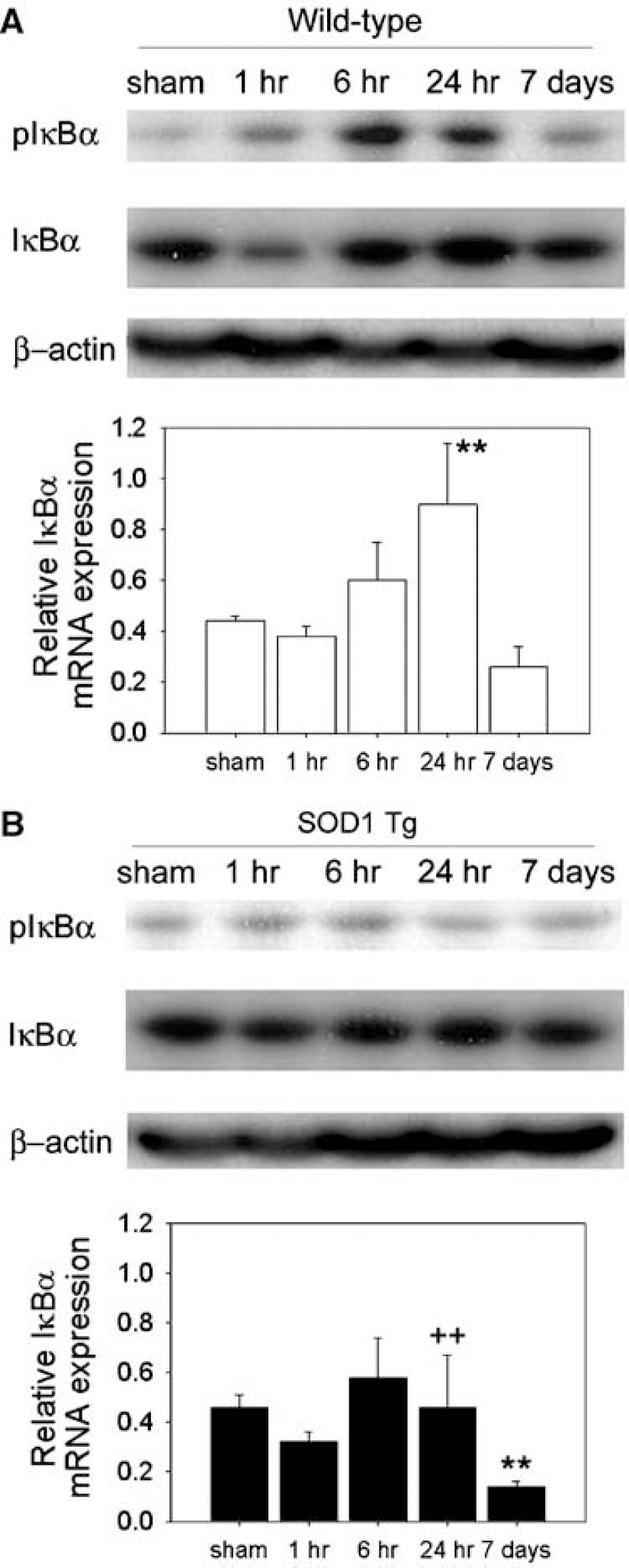
Changes in IκBα phosphorylation and degradation and IκBα mRNA after transient focal cerebral ischemia (tFCI) in the wild-type and SOD1 Tg mice. (
Superoxide Production and Ubiquitinated Protein Levels were Reduced by SOD1 Overexpression
Oxidative stress by tFCI in the wild-type mice caused a transient decrease in the IKK complex, NF-κB activation, and degradation and phosphorylation of IκBα (Ser32/36), which was subsequently ubiquitinated. However, SOD1 overexpression attenuated transient decreases in the IKK complex, NF-κB activation, and degradation and phosphorylation of IκBα (Ser32/36). This made us speculate that superoxide production and ubiquitinated protein may cause the IKK complex decrease. We determined O−2 production using HEt in both the wild-type and SOD1 Tg mice. Oxidized HEt signals were observed most strongly in the ischemic caudate 1 h after reperfusion and were reduced in the SOD1 Tg mice compared with the wild-type mice (Figure 5A). These results are consistent with previous reports that used the same tFCI model in mice (Saito et al, 2003). SOD1 prevented the early production of O−2 after tFCI. The ubiquitin-proteasome system has a central role in the selective degradation of phosphorylated or damaged proteins. Proteasomes make up to 1% of total cell protein (Hendil, 1988) and are associated with the cytoskeleton in the cytoplasm (Wójcik and DeMartino, 2003). IκB kinase-mediated phosphorylation also triggers the ubiquitination of IκBα by E3 Ligase SCFβTRCP. To elucidate the ubiquitination after tFCI, we investigated ubiquitinated protein in both the wild-type and SOD1 Tg mice in the cytoskeletal fraction after tFCI. A high elevation of ubiquitin conjugate levels in the ischemic compared with the sham animals was observed within 1 h of reperfusion in the wild-type mice (Figure 5B). The conjugated immunoreactivity remained for 24 h, but eventually decreased to control levels by 7 days. In the SOD1 Tg mice, ubiquitinated protein was increased at 1 and 6 h, but was less than in the wild-type mice (Figure 5B).
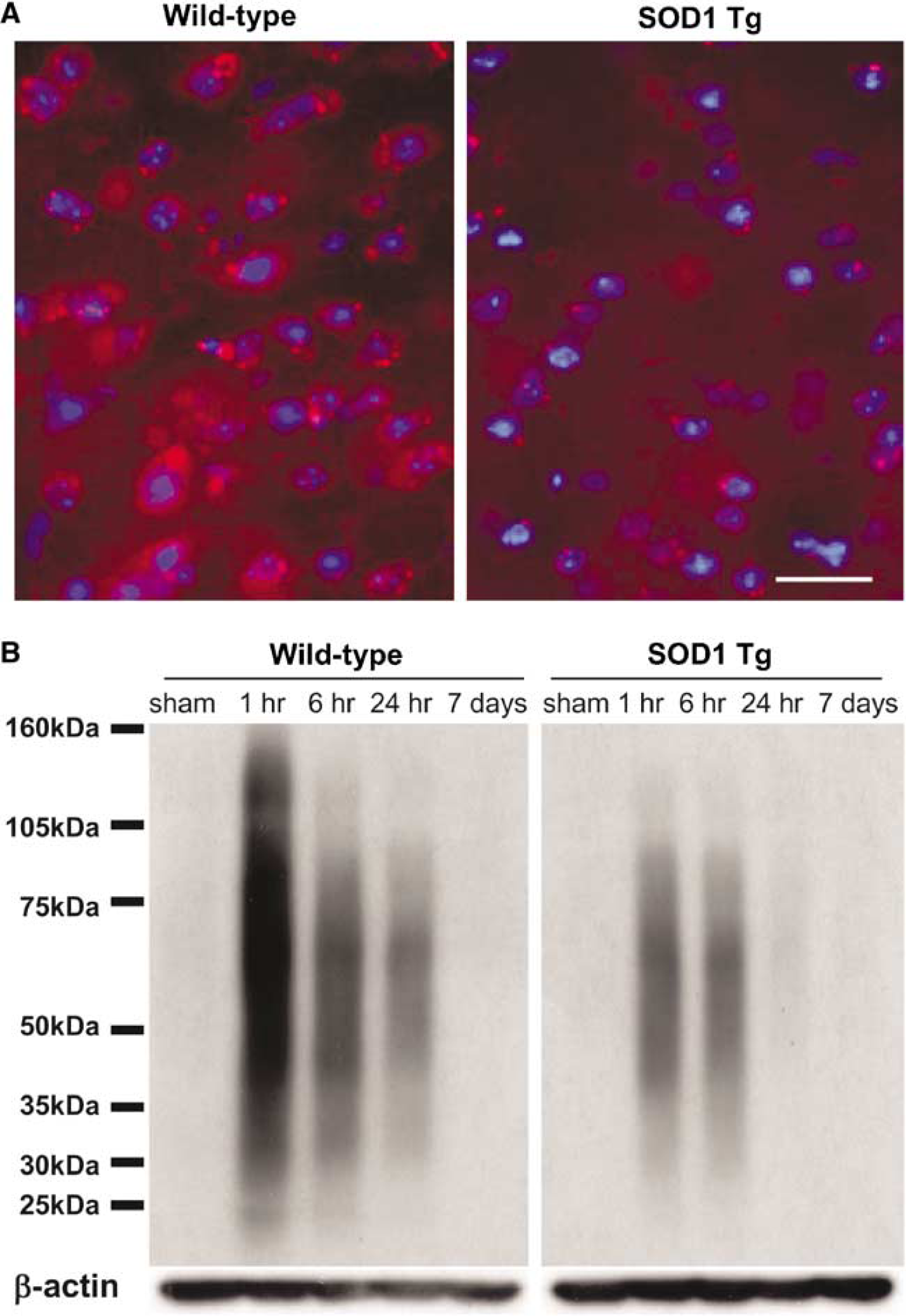
SOD1 prevents increases in superoxide production and ubiquitinated protein after transient focal cerebral ischemia (tFCI). (
Taken together, our data suggest that superoxide production and ubiquitination may directly or indirectly affect the transient decrease in the IKK complex.
Discussion
Nuclear factor-κB activity is an important cellular response that modulates the fate of cells exposed to ROS. Regulation of NF-κB in cerebral ischemia is different depending on the nature of the damaging insult, the status of oxidative stress, and the time points after injury (Denk et al, 2000). Although much effort has been made to study the molecular mechanism of NF-κB activation in cerebral ischemia, it largely remains to be identified. In this study, we demonstrated that oxidative stress causes a transient loss of the IKKα, β, and γ complex, the upstream components of NF-κB signaling, after 30 mins of MCA occlusion in the wild-type mice. Consistent with our earlier reports that NF-κB activation increased in the mouse brain after 60 mins of tFCI (Huang et al, 2001), we also found increased NF-κB DNA binding activity after 30 mins of tFCI. Transient decreases in the level of the IKKα, β, and γ complex were accompanied by NF-κB activation and IκBα phosphorylation (Ser32/36) after tFCI in the wild-type mice. Conflicting views have emerged about the decreases in the IKK complex and NF-κB activation because the seminal event in the activation of NF-κB is the phosphorylation of IκBs, which is mediated by the IKK complex. Recently, there have been several reports on the downregulation of the IKK complex in tumor necrosis factor (TNF) signaling. During apoptosis caused by TNF-R1, IKKβ was also cleaved by caspase-3 (Tang et al, 2001). The initial down-regulation of IKKα and IKKβ in TNF-α-stimulated cells appears to be caused by autophosphorylation of IKK in TNF signaling (Delhase et al, 1999). It was reported that the IKK complex is the target of the NF-κB-inhibitory TNF receptor-associated factor-1-fragment (Henkler et al, 2003). Botchkina et al (1999) suggested that rapid and progressive loss of NF-κB activity could participate in the development of TNF-induced cytotoxicity after cerebral ischemia. The IKK complex downregulated during interaction with TNF signaling. Reactive oxygen intermediates seem to be second messengers in the NF-κB signaling pathways of the nervous system (Kaltschmidt et al, 1999).
Using SOD1 Tg mice to investigate the role of ROS in the IKK complex, we found that IKKα, β, and γ mRNA and protein levels were not affected by tFCI. SOD1 also prevented NF-κB activation and IκBα degradation after tFCI. Superoxide production and ubiquitinated protein in the SOD1 Tg mice were also lower than in the wild-type mice after tFCI. This finding raises the possibility that transient loss of the IKKα, β, and γ complex after tFCI is mediated by ROS. Nuclear factor-κB activity might be regulated by the redox regulation system (Oka et al, 2000). IKKα and β contain a cysteine residue at position 179 within their activation loop. Replacement of cys179 with alanine does not modify IKKβ activation by NF-κB-inducing kinase (Rossi et al, 2000). Regulation of the sulfhydryl group of cys179 in IKKα and β by oxidative stress can change conformation of the IKK complex and subsequent NF-κB signaling (Schoonbroodt and Piette, 2000). Recently, it was also shown that oxidative stress can activate p90rsk via Fyn and Ras (Abe et al, 2000). This stimulation occurs in various cell lines and is extensive, rapid, and transient. p90rsk could collaborate or synergize with the IKK complex in NF-κB activation (Schoonbroodt and Piette, 2000). The ubiquitin-proteasome system for degradation of the most short-lived, damaged, or misfolded proteins, as well as long-lived proteins, is important in the regulation of a number of key biological regulatory mechanisms (Palombella et al, 1994; Glickman and Ciechanover, 2002). In cerebral ischemia in vivo, multiubiquitin chains persistently increased in the area where neurons are destined to die, and accumulation of multiubiquitin is caused by proteasomal dysfunction. The increase in inappropriate multiubiquitin proteins may aggravate apoptotic neuronal cell death after cerebral ischemia (Ide et al, 1999; Asai et al, 2002). Phosphorylated IκBα also mediated by the IKK complex is ubiquitinated, which targets it for degradation by the 26S proteasome. Nuclear factor-κB inducers target IKKβ for conjugation to ubiquitin in mammalian cells. Carter et al (2003) proposed that T loop phosphorylation at Ser177/Ser181 generates a conditional ubiquitin-targeting signal in IKKβ by a post-translational mechanism. Increases in accumulation of multiubiquitin after 30 mins of tFCI may show the ubiquitination of the IKK complex. We also showed that accumulation of multiubiquitin was prevented by SOD1 after tFCI. To substantiate the idea that the central IKK complex after tFCI is oxidant-responsive or redox-regulated, continued study is required. Because the IKK complex is a unique bottleneck through which all the disparate stimuli that activate NF-κB must pass, it would be an important target in the study of the NF-κB mechanism.
We would argue that the changes in IKK and IκB in the ischemic brain seen in mice overexpressing SOD1 may merely reflect the lesser degree of ischemic injury. In our stroke model, the expression of ischemic damage generally occurred beginning at 6 h, and the lesion matured much later (i.e., 24 to 72 h). Thus, we believe that the early changes in IKKα, β, and γ and NF-κB activity as early as 1 h after reperfusion do not reflect the effect of lesser ischemic damage. However, in stroke mice that overexpressed SOD1, all these early changes were altered because of less oxidative stress than generally occurs during reperfusion. We thus conclude that the changes in NF-κB upstream signaling are altered by oxidative stress and that such alterations may be one of many factors that affect the stroke outcomes rather than the other way around.
In the current study, we provide the first evidence that oxidative stress is involved in the transient decrease of the IKK complex in cerebral ischemia. Our results suggest that increased ROS after tFCI are key elements that downregulate the IKK complex through redox-sensitive modification of proteins, not only directly but also indirectly. We conclude that this study provides insights into the NF-κB upstream mechanisms triggered by oxidative stress during cerebral ischemia and reperfusion.
Footnotes
Acknowledgements
The authors thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.