
Abstract
Neuroprotective properties of ketosis may be related to the upregulation of hypoxia inducible factor (HIF)-1α, a primary constituent associated with hypoxic angiogenesis and a regulator of neuroprotective responses. The rationale that the utilization of ketones by the brain results in elevation of intracellular succinate, a known inhibitor of prolyl hydroxylase (the enzyme responsible for the degradation of HIF-1α) was deemed as a potential mechanism of ketosis on the upregulation of HIF-1α. The neuroprotective effect of diet-induced ketosis (3 weeks of feeding a ketogenic diet), as pretreatment, on infarct volume, after reversible middle cerebral artery occlusion (MCAO), and the upregulation of HIF-1α were investigated. The effect of β-hydroxybutyrate (BHB), as a pretreatment, via intraventricular infusion (4 days of infusion before stroke) was also investigated following MCAO. Levels of HIF-1α and Bcl-2 (anti-apoptotic protein) proteins and succinate content were measured. A 55% or 70% reduction in infarct volume was observed with BHB infusion or diet-induced ketosis, respectively. The levels of HIF-1α and Bcl-2 proteins increased threefold with diet-induced ketosis; BHB infusions also resulted in increases in these proteins. As hypothesized, succinate content increased by 55% with diet-induced ketosis and fourfold with BHB infusion. In conclusion, the biochemical link between ketosis and the stabilization of HIF-1α is through the elevation of succinate, and both HIF-1α stabilization and Bcl-2 upregulation play a role in ketone-induced neuroprotection in the brain.
Introduction
Ketone bodies, such as R-β-hydroxybutyrate (BHB) and acetoacetate (AcAc), are alternate energy substrates to glucose and are utilized by the brain and most other tissues especially during early development and under conditions of reduced glucose availability, such as starvation, fasting, heavy exercise, and strict caloric intake of a highfat, low-carbohydrate diet (Freeman et al, 1998; Melo et al, 2006; Veech, 2004; Nehlig, 2004). The relationship between ketone body metabolism by the brain and neuroprotection continues to be investigated, as there are many proposed mechanisms that ketones act through (Prins, 2008). In a rodent model of middle cerebral artery occlusion, infarct volume was reported to be reduced with ketosis as a result of inhibition of lipid peroxidation and hence reduction of reperfusion injury, as well as an overall improvement of cerebral energy metabolism (Suzuki et al, 2002). Neuroprotection by ketones has been proposed to be linked to glucose metabolism through the downregulation of glycolysis, possibly through decreased phosphofructokinase activity and, thus, a reduction in lactic acid accumulation during ischemia (Gueldry et al, 1990). However, local cerebral glucose utilization measured by the [14C]2-deoxy-D-glucose method in conscious hyperketonemic rats (induced by fasting or exogenous infusion) showed no change in the cerebral metabolic rate for glucose (Corddry et al, 1982).
Ketosis has been considered, and in some cases utilized, as a therapeutic strategy for the treatment of hypoglycemia, seizure disorders (induction of ketosis by ketogenic diet), and as an alternative to high-lipid parenteral and enteral feedings (Desrochers et al, 1995; Freeman et al, 1998; Yudkoff et al, 2007). Since hyperglycemia has been known to exacerbate brain ischemic damage (Parsons et al, 2002; Tsuda, 2006), one could hypothesize to use ketosis as a treatment to counteract the hyperglycemia. Ketosis has also been considered to improve hypoxic tolerance by improving the metabolic energy state that results from an imbalance in glucose metabolism and energy insufficiency (Suzuki et al, 2002). Treatment with ketone body precursors, such as 1,3-butanediol, has shown promising results with respect to diminished brain damage because of ischemia and/or reperfusion injury (Gueldry et al, 1990; Sims and Heward, 1994).
A potential benefit of ketone bodies toward improving overall physical and cognitive performance as well as protection from oxidative stress is thought to be linked to improved metabolic efficiency (Maalouf et al, 2007; Veech, 2004). Ketone bodies are suggested to have a beneficial role in mitochondrial energy metabolism through the stabilization of redox, as well as nonoxidative, metabolism, such as through the activation of survival pathways (Guzman and Blazquez, 2004; Kowaltowski and Fiskum, 2005; Sullivan et al, 2004; Suzuki et al, 2002; Veech, 2004; for review, Prins, 2008). However, the biochemical and molecular mechanisms that link the metabolic effects of ketosis with neuroprotection remain to be discerned. In a recent review, neuroprotection by ketosis was discussed and at least 17 key findings associated with the use of ketone therapy in various central nervous system injury models were presented, which suggests that ketones act through more than a single mechanism, but most likely through multiple mechanisms (Prins, 2008).
A possible link between ketosis, neuroprotection, and angiogenesis is through hypoxia inducible factor (HIF)-1α (Chavez et al, 2000), which is a transcription factor related to the regulation of energy metabolism and is known to accumulate during hypoxia (Semenza, 2004). Hypoxia inducible factor-1α is thought to be one of the most crucial signaling molecules that results in upregulating survival pathways associated with oxygen deprivation, as well as mediating neuroprotective responses associated with metabolic compromise (Freeman and Barone, 2005; Soucek et al, 2003). Our hypothesis was that the neuroprotective properties of ketosis are through the upregulation of HIF-1α. The rationale was developed on the basis that the utilization of ketones results in the elevation of intracellular succinate. Increased succinate has been shown to induce HIF-1α through the inhibition of prolyl hydroxylase (PHD; EC 1.14.11), the enzyme responsible for the degradation of HIF (Myllyla et al, 1977).
Results from our previous study suggest that the link between ketosis and neuroprotection is related to angiogenesis (Puchowicz et al, 2007). The current study establishes that a primary player associated with hypoxic angiogenesis, HIF-1α, is upregulated in ketotic brains of normoxic rats either fed a ketogenic diet or intraventricularly infused with the ketone body BHB. From these data, together with our understanding of the regulation of HIF-1α, we explored a possible biochemical explanation that relates ketosis, angiogenesis, and neuroprotection.
In this study, we explored a potential biochemical mechanism related to the effects of ketosis on neuroprotection. We hypothesized that ketosis ameliorates the damage caused by reperfusion injury after stroke induced by MCAO in rat. Based on the rationale that cerebral utilization of ketone bodies has beneficial effects on ischemic response, diet-induced ketosis or brains intraventricularly infused with Na-BHB as a pretreatment on infarct volume after reversible MCAO were studied. Succinate content and HIF-1α response in ketotic rat brain were also studied.
Materials and methods
Animal Preparation and Feeding Conditions
All procedures were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University. Male Wistar rats, 28 days old and weighing 110 g, were allowed to acclimate in the CWRU animal facility for 1 week before being used in experiments. The experimental design consisted of two studies.
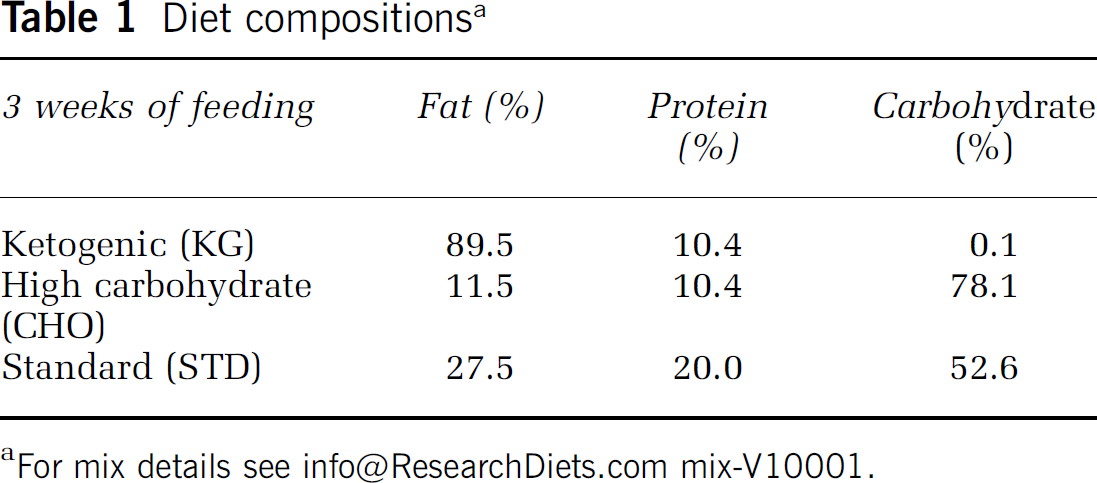
We developed a diet-induced rat model of chronic ketosis by feeding the KG diet to young adult rats. In this model, we have shown that blood ketone levels are similar to what are measured in humans who are on a 3-day fast or are fed a KG diet (Puchowicz et al, 2007). The advantages of inducing ketosis by feeding a KG diet is that ketosis can be sustained for days and months rather than hours (Desrochers et al, 1995), as with exogenous infusions of ketones or precursors of ketone bodies. This approach also eliminates the complications often encountered with exogenous administration of ketones as sodium salts or acids, such as sodium overload or vascular irritation.
Experimental Setup for Middle Cerebral Artery Occlusion and Molecular Analyses
Rats ketotic by diet or substrate-infused, and their matched controls (nonketotic diets or NaCl-infused), were anesthetized with 2% halothane/O2/N2O and MCAO was performed using a monofilament model (Lust et al, 2002), occluded for 2 h; rats were killed at 24 h of reperfusion and the infarct volumes determined (see below for description). The MCAO infarct and edema volumes were measured in the diet-fed and infused groups. The analysis of proteins, HIF-1α and Bcl-2 (by western blotting), and mRNA expression of PHD, as well as the succinate content (nmol/mg wet weight; gas chromatography—mass spectrometry), required subgroup of rats that were ketotic by diet or substrate-infused, and in which MCAO was not performed.
Infarct Volumes and Edema Measurements
The infarct volumes after focal cerebral ischemia (by MCAO) were determined by a standard procedure as previously described (Duverger and MacKenzie, 1988). Rats were deeply anesthetized and transcardially perfused with ice-cold heparinized PBS (phosphate-buffered saline, pH 7.4) to remove excess blood. Brains were removed, sectioned (20 μm coronal sections) on ice using cryostat blades, and stained histochemically with TTC (2,3,5-triphenyltetrazolium chloride); TTC staining measures tissue viability used to evaluate infarct size. As ischemic regions tend to expand on mounting the tissue on the slide (an approximation of edema), the infarct area was determined by measuring the total contralateral hemisphere and subtracting the normal tissue area on the ipsilateral hemisphere. This approach minimized the artifact of swelling and yielded a more accurate assessment of the actual infarct areas (Swanson and Sharpe, 1994). The formula for infarct volume (Vi
HIF-1α and Bcl-2 Protein Levels by Western Blot Analysis
In another set of animals (non-MCAO), HIF-1α and Bcl-2 protein contents were studied in the diet-induced ketotic rat brain or brains infused with substrate using western blot analysis. Animals were deeply anesthetized with halothane/O2/N2O and decapitated. The brains were quickly removed, frozen in liquid nitrogen, and stored at −80°C. Cortical (frontal or parietal regions, approximately 50 to 100 mg wet weight) samples were dissected from either the right or left hemisphere and homogenized in ice-cold buffer: 250 ml of buffer A (20 mmol/L HEPES, 1.5 mmol/L MgCl2, 0.2mmol/L EDTA, 100 mmol/L NaCl, 0.5 mmol/L PMSF, 1 μg/ml leupeptin and 0.2 mmol/L DTT, pH 7.4). Additional NaCl was added to the homogenate to obtain a final concentration of 0.45 mol/L NaCl. The homogenate was mixed and then placed in a centrifuge for 30 min at 14,000 r.p.m. at 4°C. The supernatant was collected (lysate) and measured so that an equal volume of buffer B (buffer A plus 40% glycerol; vol/vol) was added before being stored at —80°C. Lysate samples were then analyzed for protein concentration using the Bradford Assay method before electrophoresis for western blotting (Agani et al, 2002).
Equal quantities of protein (100 μg of the lysate) were boiled with gel-loading buffer for 5 min before loading onto a 10% SDS-polyacrylamide gel. Separated proteins were transferred to nitrocellulose membranes using a Bio-Rad semi-dry transfer apparatus (Bio-Rad Laboratories, Hercules, CA, USA). Blots were blocked overnight in 5% skim milk powder in PBS—Tween 20 at 4°C, and probed with anti-Bcl-2 and anti-β-actin (both from Santa Cruz Biotechnologies, Santa Cruz, CA, USA), or anti-HIF-1α, a generous gift from Dr F Agani (Department of Anatomy, Case Western Reserve University), with 3% bovine serum albumin in PBS—Tween 20. Blots with bound primary antibody were incubated with peroxidase-labeled secondary antibodies (Jackson Laboratories, West Grove, PA, USA). Detection of the bound secondary antibody employed the Pierce (Rockford, IL, USA) ECL western blotting detection reagents. Densitometry was performed with SigmaScan Pro5™ image analysis software, and values were normalized to the β-actin content and expressed as relative intensity.
Prolyl Hydroxylase mRNA Expression by Real-Time PCR
Prolyl hydroxylase mRNA expression was studied in the diet-induced ketotic rat brain (non-MCAO) by real-time PCR. Cortical (frontal or parietal regions, approximately 50 to 100 mg wet weight) samples were freshly dissected (from either right or left hemisphere), quickly homogenized in guanidine-isothiocyanate-containing lysis buffer, and RNA was isolated using the Quiagen RNeasy kit according to the manufacturer's instructions (Quiagen, Valencia, CA, USA). Five micrograms of total RNA was treated with DNAse I (Gibco, Carlsbad, CA, USA), and then first-strand cDNA synthesis was performed using a reverse transcription reaction, using oligo d(T)15 (Promega, Madison, WI, USA) and superscript II reverse transcriptase (Life Technologies, Rockville, MD, USA). PCR amplification and detection were performed using Bio-Rad i-Cycler™ detection system (Bio-Rad Laboratories) in a total volumeof 25 μL containing 50 mmol/L KCl, 20 mmol/L Tris—HCl (pH 8.4), 0.2 mmol/L of each dNTP, 25U/mL iTaq DNA polymerase, 3 mmol/L MgCl2, SYBR Green I, 10 nmol/L fluorescein, and stabilizers. Each reaction also contained 300 nmol/L primer. Primer sequences for PHD were 5‘-TGTTGGATCAGAACCACGACGGTC-3’ (forward) and 5‘-GAGTCATCCACCGTCCATTT-3’ (reverse). Each amplification was performed in triplicate, using the following conditions: 3 mins at 95°C, 30 secs at 95°C, and 30 secs at 60°C through 40 cycles, followed by 2 temperature cycles (30 secs at 72°C and 1 min at 60°C). A melt curve protocol (70 cycles at 60°C to 95°C, with an increment of 0.5°C/cycle) was performed after each amplification to assess the purity of the product. In addition, amplified products for each primer set were analyzed using electrophoresis to confirm product size and purity. Relative expression was calculated by comparing the cycle thresholds for the genes of interest with the cycle thresholds for β-actin mRNA.
Succinate Content
Succinate content (nmol/mg wet weight) was analyzed from extracts prepared from quick-frozen (using liquid nitrogen and storing at −80°C) cortical brains. Samples were collected from the frontal region (right or left hemisphere, weighing approximately 50 mg wet weight) from either diet-induced ketotic rat brain or brains infused with substrate. Frozen samples were spiked with internal standard [U-13C4]succinate (Isotec, Miamisburg, OH, USA), acidified with sulfosalicylic acid, and homogenized with choloroform/methanol (2:1) solvent mixture at −25°C using a Polytron homogenizer. Extracts were then analyzed using gas chromatography—mass spectrometry on an Agilent 5973 mass spectrometer, equipped with an Agilent 6890 gas chromatograph and an HP-5MS 5% phenyl methyl siloxane fused silica capillary column (60 m, 250 mm inner diameter, 0.25 μm film thickness). Sample preparation and gas chromatography—mass spectrometry procedures were performed as previously described (Yang et al, 2006).
Statistical Analysis
Statistical analysis was performed on the data collected from the diet groups, KG, CHO, and STD, and the substrate-infused groups, Na-BHB, 3-NPA, Na-propionate, and NaCl; the numbers (
Results
Infarct and Edema Volumes
Infarct volumes were measured in rats fed the three diets: STD (
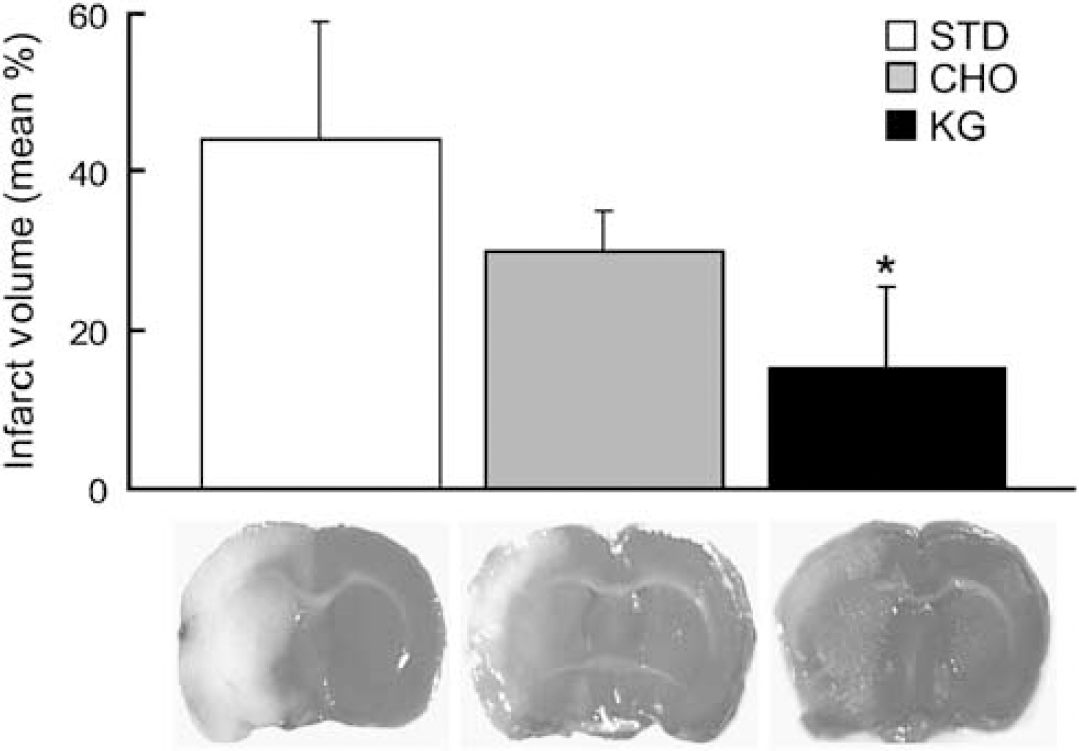
Decreased infarct volume in diet-induced ketotic rat brain. Infarct volumes as determined by TTC staining in the diet-fed groups (STD-control, CHO, KG) are presented as the mean percentage infarct volume of the ipsilateral hemisphere. Measurements were blinded and randomized. *
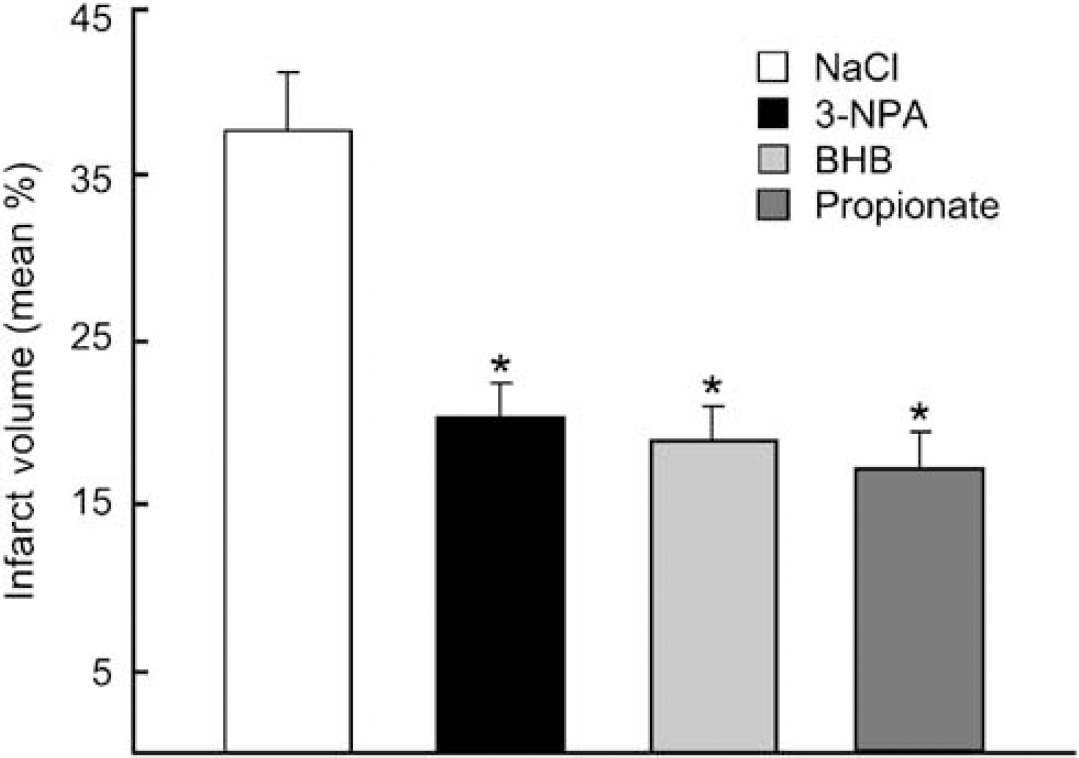
Decreased infarct volumes in substrate-infused rat brain. Infarct volumes as determined by TTC staining in the substrate-infused groups (NaCl-control, BHB, 3-NPA, Propionate) are presented as the mean percentage infarct volume of the ipsilateral hemisphere. Measurements were blinded and randomized. *
HIF-1α and Bcl-2 Protein Levels and Prolyl Hydoxylase mRNA Content
The levels of HIF-1α protein were elevated in the rats fed the KG diet or BHB-infused, when compared with matched controls (STD diet or NaCl-infused;
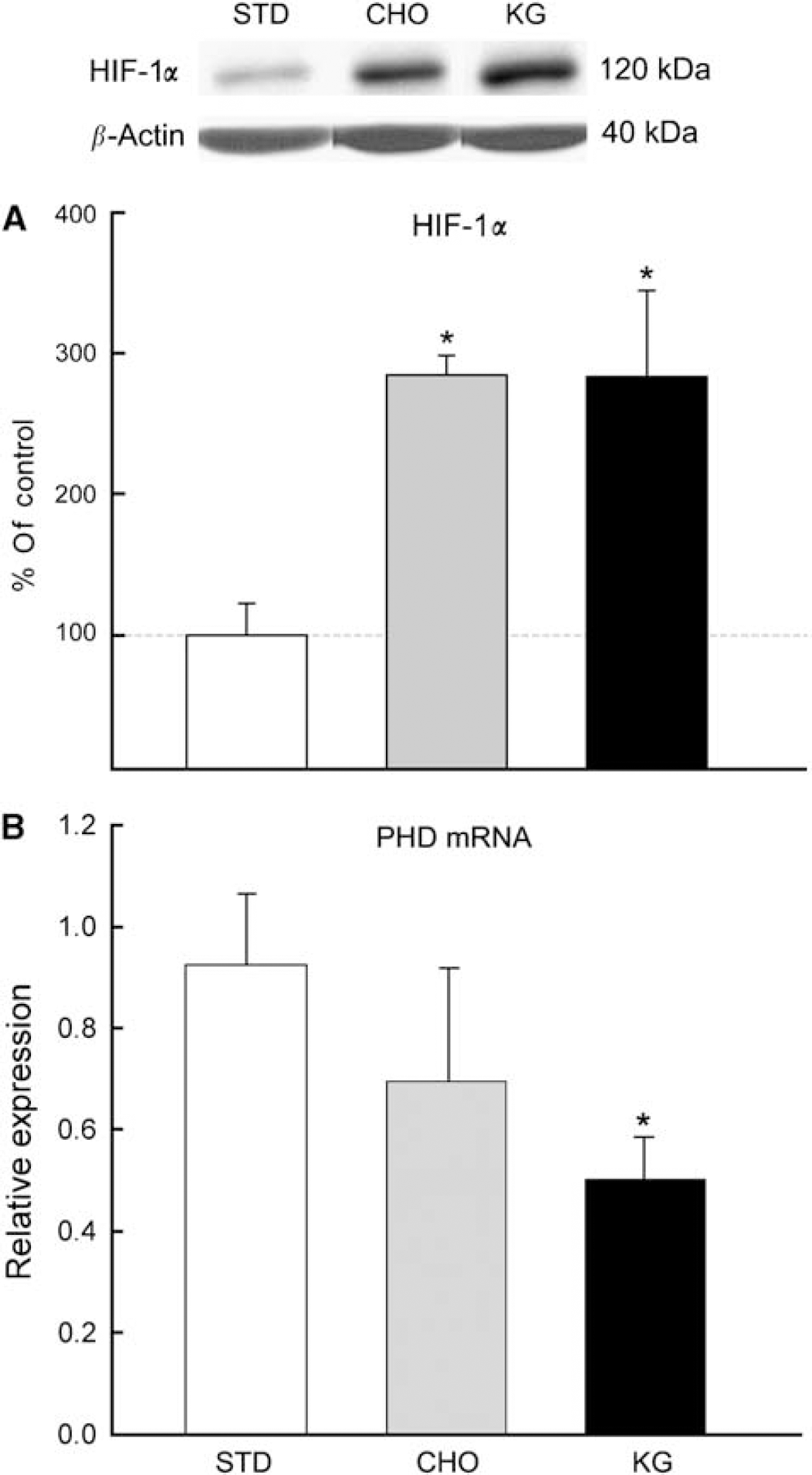
Level of HIF1-α protein and PHD mRNA expression in diet-induced ketotic rat brain. Western blot and densitometric analysis revealed a threefold increase in the HIF1-α protein levels in the CHO and KG diet groups (
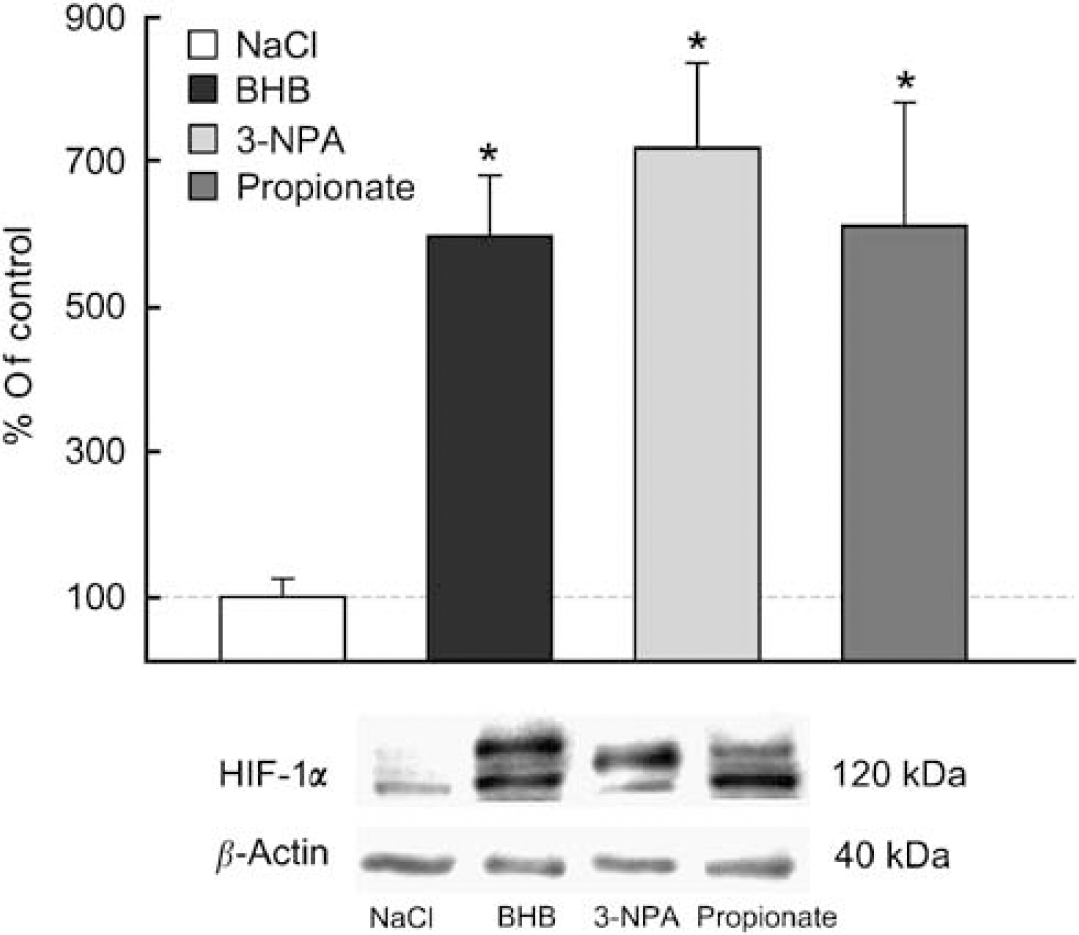
Upregulation of HIF-1α protein in substrate-infused rat brain. Western blot and densitometric analysis revealed a fivefold increase in HIF1-α protein levels in each of the intraventricularly substrate-infused rats (BHB, 3-NPA, Propionate) compared with NaCl-control. Data are expressed as percentage of NaCl-control. *
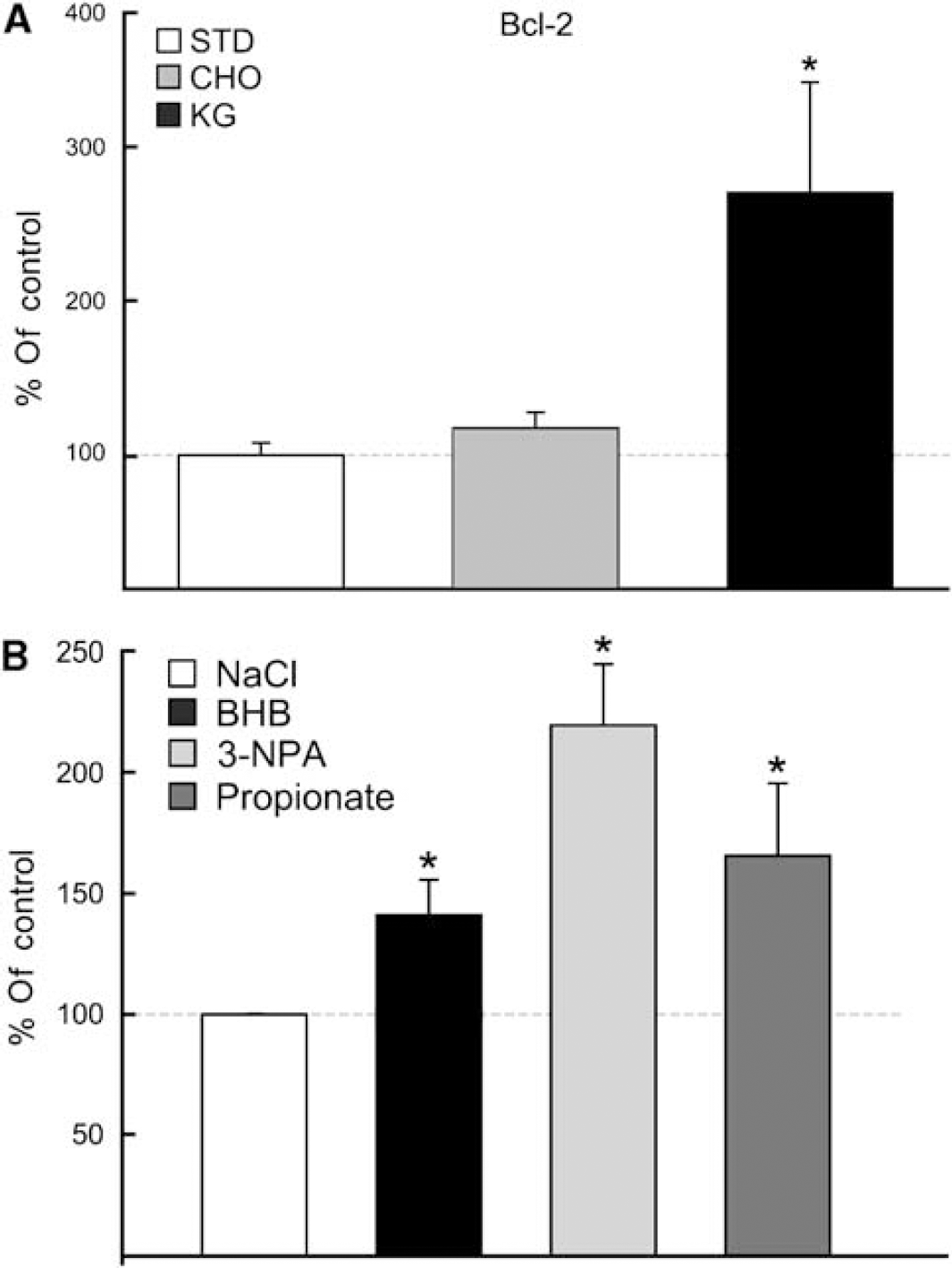
Upregulation of Bcl-2 protein. Western blot and densitometric analysis revealed a threefold increase in Bcl-2 protein levels with diet-induced ketosis and no change with the CHO diet, when compared with the STD-control group (
As observed with the diet-fed groups, western blot analysis revealed an upregulation of Bcl-2 in each of the substrate-infused rats (BHB, 3-NPA, and propionate) (Figure 5B). Bcl-2 protein increased by 50% in the BHB-infused group compared with the NaCl-infused controls. A similar increase was measured in the propionate-infused group. The greatest induction of Bcl-2 was observed in the 3-NPA group (twofold relative to the NaCl-infused controls).
Succinate Content
Succinate content (nmol/mg wet weight) was measured in the diet-fed groups (STD, KG) and substrate-infused rat brains (NaCl, BHB, 3-NPA, and propionate), and in both conditions (diet-induced ketosis and BHB-infused) succinate was significantly elevated compared with the STD diet or NaCl controls. As expected, the succinate content in 3-NPA and propionate-infused groups was also significantly increased (similar to what was observed with the KG diet and BHB-infused). These two substrate-infused groups were added as positive controls, as these compounds are known to result in increased succinate levels. Gas chromatography-mass spectrometry analysis revealed that in brain tissue preparations for succinate concentrations, the KG diet-fed group was significantly elevated by 1.6-fold (0.28 ± 0.08) compared with the STD diet group (0.18 ± 0.02) (Goldberg et al, 1966). The succinate levels measured in the substrate-infused groups were similar to what were measured in the KG diet-fed group, as there was a fivefold increase in the BHB- and propionate-infused and a threefold increase in the 3-NPA groups, when compared with the NaCl-infused control group (0.06 ± 0.04) (Figure 6).
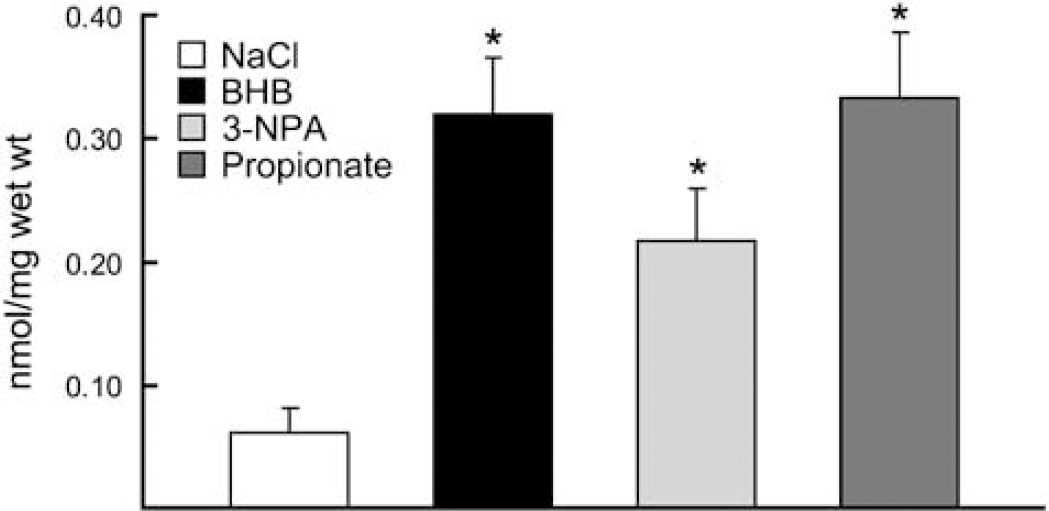
Succinate content in the substrate-infused groups. Gas chromatography-mass spectrometry analysis of succinate concentrations in BHB substrate-infused brains were significantly elevated fivefold compared with the NaCl-control and similar to the positive controls (3-NPA and Propionate, as these toxic compounds are known to result in increased succinate levels). Data are expressed as nmol/mg wet weight. *
Discussion
The role of ketone bodies as alternate energy substrates in the brain has prompted investigators to explore their potential use as a therapeutic treatment modality against neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Freidreich's ataxia, amyotrophic lateral sclerosis, and against ischemic-reperfusion injury (Kashiwaya et al, 2000; Zhao et al, 2006). Although the associated mechanisms remain unknown, it is hypothesized that ketone bodies play a neuroprotective role by improving metabolic efficiency, sparing of glucose oxidation, and degradation of muscle-derived amino acids, and, in some neurodegenerative conditions, protect against glutamate cytotoxicity. In this study, we report a significant reduction in infarct volume after MCAO in diet-induced ketotic rats or rats intraventricularly infused with Na-BHB.
We have previously shown an increase in brain capillary density in our 3-week diet-induced rat model of ketosis (Puchowicz et al, 2007), similar to what has been observed in nonketotic rat brain after 3 weeks of hypobaric-hypoxic-induced angiogenesis (Pichiule and LaManna, 2002). Angiogenesis has been recognized to play an important role associated with improved outcome from stroke (Slevin et al, 2006). The data from this study resulted in a significant elevation in succinate levels in ketotic rat brain. Ketone body metabolism has been known to result in increased levels of citric acid cycle intermediates, such as citrate and succinate, and thus we hypothesized that the metabolic side of HIF-1α regulation via ketosis is through product inhibition of PHD by succinate (Myllyla et al, 1977; Selak et al, 2005), an intermediate of energy metabolism (see Figure 7). Succinate is produced as an intermediate of the citric acid cycle (mitochondria) through a series of reactions that uses 2-oxoglutarate (also known as 2-ketoglutarate) and succinyl-CoA as substrates, and the enzymes 2-oxoglutarate dehydrogenase complex and succinyl-CoA synthetase, respectively. In the cytosol, succinate is a product of the conversion of 2-oxoglutarate by the PHD enzyme when oxygen is not limiting.
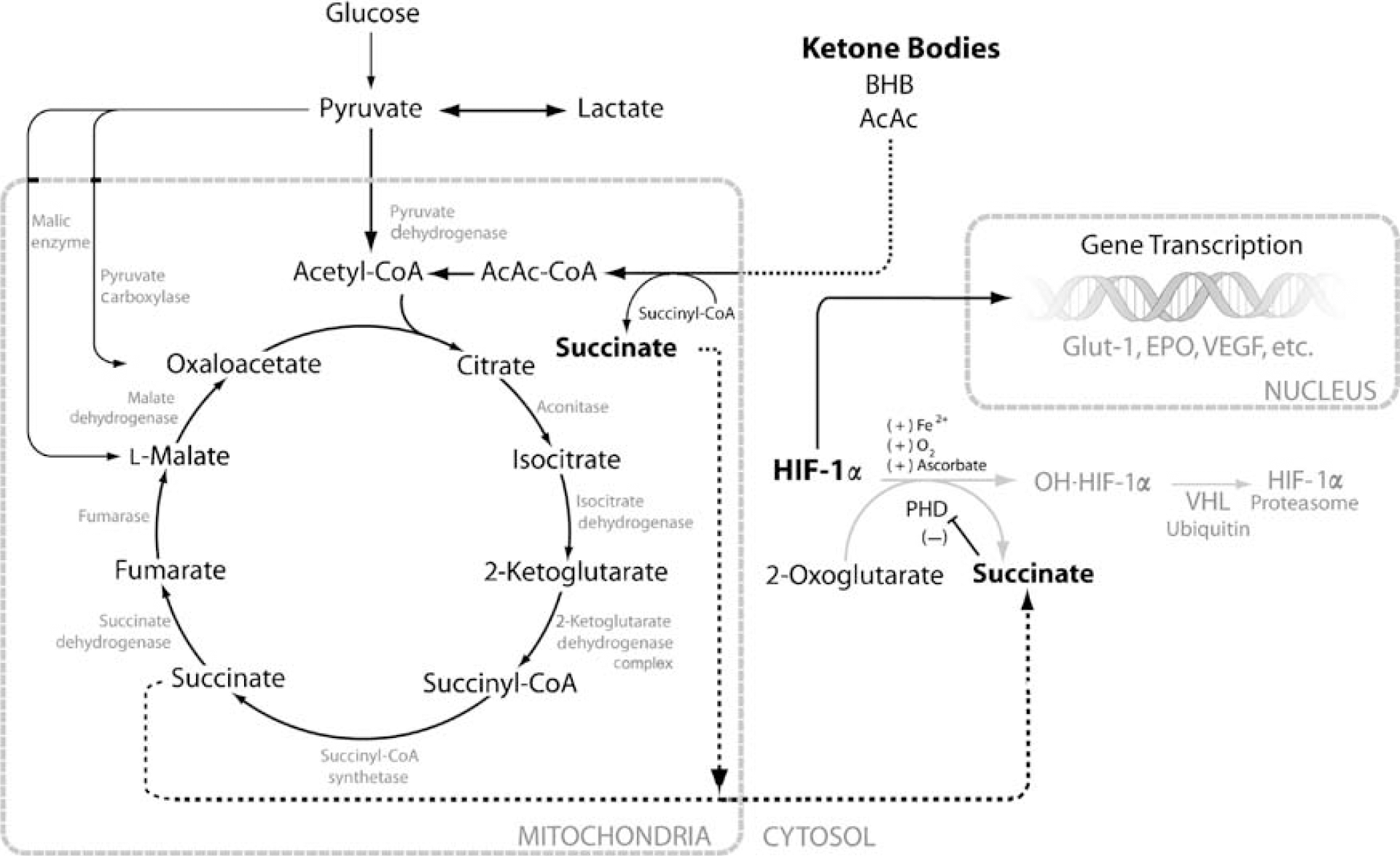
Metabolic pathway of ketone body utilization and the relationship with the stabilization of HIF-1α. Our scheme of the neuroprotective properties of ketone bodies relates ketone body metabolism to the stabilization of HIF-1α through product inhibition of prolyl hydoxylase (PHD) by succinate as follows: glucose (via glycolysis) and ketone bodies (acetoacetate and β-hydroxybutyrate; AcAc, BHB) in brain mitochondria enter the citric acid cycle as acetyl-CoA residues and through a condensation reaction with oxaloacetate, catalyzed by citrate synthase, form citrate. Succinate is generated by the catabolism of acetyl-CoA via the citric acid cycle and through the activation of AcAc to AcAc-CoA by the enzyme succinyl-CoA-AcAc-CoA transferase (β-oxoacid-CoA-transferase). Activation of AcAc to AcAc-CoA requires succinyl-CoA as the CoA donor and the resultant product is succinate. The proposed mechanism for the stabilization of HIF-1α is shown to occur through the accumulation of mitochondrial succinate. With ketone body utilization, mitochondrial succinate is elevated; maintaining the succinate mitochondrial pool would require transport of succinate out of the mitochondria via dicarboxylic acid transporter system into the cytosol. Elevation of succinate in the cytosol results in the stabilization of HIF-1α through the product inhibition of PHD. During conditions when succinate does not accumulate, PHD is the enzyme that catalyzes the reaction of HIF-1α to the hydroxylated form (OH-HIF-1α) and undergoes proteasome degradation. Hydroxylation also requires 2-oxoglutarate (also known as 2-ketoglutarate; refers to the cytosolic substrate, whereas 2-ketoglutarate notation in the citric acid cycle refers to the mitochondrial substrate) as a substrate and generates succinate as a coproduct. The resultant increase in HIF-1α stimulates the production of a vasogenic activator (vascular endothelial growth factor, VEGF) and erythropoietin (EPO), and also stimulates survival pathways, through the activation of related target genes, including Bcl-2.
Succinate and 2-oxoglutarate can be transported across the mitochondrial membranes via specific substrate transporter systems, which stabilize a ‘redox’ balance between forward and reverse reactions. An acute shift in redox state results in biochemical perturbations, which act as signal transducers. Our rationale is that HIF-1α is stabilized through inhibition of the cytosolic enzyme PHD via succinate, and thus inhibits the forward biochemical reaction that requires 2-oxoglutarate (Acker and Acker, 2004; Dalgard et al, 2004). Consistent with our hypothesis, a significant decrease in PHD mRNA expression in the ketotic rat brain was also observed. Others have shown similar results where HIF-1α accumulation without hypoxic stimulation was as a result of the inhibition of PHD through the chelation of iron (Myllyla et al, 1977; Park et al, 2007). These and other studies have investigated the potential role of HIF-1α in neuroprotective preconditioning (Freeman and Barone, 2005; Soucek et al, 2003).
As HIF-1α has been described to regulate genes involved with energy metabolism and antiapoptosis (Dery et al, 2005; Selak et al, 2005; Soucek et al, 2003), we also tested the possibility that this transcription factor is related to neuroprotection through the upregulation of Bcl-2 protein, an antiapoptotic protein. This multifunctional protein is a key regulator of apoptosis, which either results in cell death or survival. It protects against apoptosis (antiapoptotic mechanism) and necrosis through the regulation of mitochondrial redox or stabilization of the mitochondrial membrane by interfering with the activation of proapoptotic proteins, such as cytochrome
Elevated HIF-1α levels in the brain of the CHO diet group were similar to what were found in the ketotic group; this was unexpected and cannot be explained by this study. The purpose of feeding the CHO diet was to add an additional control to the KG diet, as we believe that the standard rodent chow (Teklad) was not a sufficient control diet, as it was manufactured by a different company. However, in this diet group there was no neuroprotection after MCAO or upregulation of Bcl-2, and thus we concluded that the upregulation of HIF-1α in the CHO group was not the only regulatory mechanism responsible for neuroprotection. These data show that increased levels of HIF-1α alone do not result in neuroprotection and that neuroprotection by ketosis may require both stabilization of HIF-1α and upregulation of Bcl-2.
To further investigate if elevated HIF-1α and succinate levels in the brain of ketotic rats chronically fed a KG diet were not a result of an unknown systemic effect of the diet, we administered ketones locally by intraventricular infusion of Na-BHB in the brains of nonketotic rats and found the same degree of neuroprotection as measured by decreased infarct volume after MCAO. Increased succinate content and HIF-1α and Bcl-2 protein levels were also observed. These results were consistent with the results observed with ketosis induced by diet.
One mechanism explaining the therapeutic effects of ketones is through the partitioning of glucose away from oxidative metabolism toward the replenishment of the citric acid cycle intermediates (
We recognized that multiple mechanisms exist for the action of ketosis. In this study, the biochemical mechanism associated with neuroprotection, upregulation of HIF-1α and succinate, was further explored in another group of rats in which propionate or 3-NPA was infused intraventricularly. These compounds are known to increase intracellular succinate and were used to test if an increase in succinate level without ketosis results in succinate-linked neuroprotection after MCAO, and were not studied as treatment strategies against reperfusion injury for they are known to be toxic. We concluded that the resultant neuroprotection with ketosis (induced by diet or locally by intraventricular infusion of BHB), together with induction of HIF-1α and Bcl-2, was through the purported action of succinate. These data suggest a link between ketosis and neuroprotection through the metabolic side of HIF. Since ketosis persisted both during and after ischemia, it was unclear at what stage ketosis was neuroprotective.
Conclusion
Ketosis (induced by diet or by local intraventricular infusion of Na-BHB) significantly reduced infarct volumes by approximately 55% greater than those of control after 2 h of ischemia and 24 h of reflow. These data indicate that (i) the biochemical link between ketosis and the stabilization of HIF-1α is through the elevation of succinate and (ii) both HIF-1α stabilization and Bcl-2 upregulation play a role in ketone-induced neuroprotection in the brain. This study establishes that ketosis as pretreatment is neuroprotective but does not ascertain whether the effectiveness occurs during ischemia and/or reperfusion. Although there is a clinical relevance of the use of ketosis (induced by diet or infusion) as a potential treatment for stroke, the use of the KG diet as a pretreatment for those at risk for stroke has not been established. The transcriptional regulation of HIF-1α through the succinate-dependent mechanism remains to be explored. Pharmacological treatment regimes (preconditioning or after ischemia) that explore the use of ketones (or ketone body analogues) as a strategy toward increased neuroprotection are needed.
Footnotes
Acknowledgements
We thank Constantinos P Tsipis for technical efforts and assistance in the preparation of this paper and Dr H Brunengraber (Department of Nutrition, Case Western Reserve University) and his staff, especially Lili Yang, for their generous support with the analysis of succinate content and use of the gas chromatograph—mass spectrometer. We also thank Dr Faton Agani (Department of Anatomy, Case Western Reserve University) for his generous gift of anti-HIF-1α.