
Abstract
Traumatic brain injury (TBI) causes both direct and delayed tissue damage. The latter is associated with secondary biochemical changes such as cell cycle activation, which leads to neuronal death, inflammation, and glial scarring. Flavopiridol—a cyclin-dependent kinase (CDK) inhibitor that is neither specific nor selective—is neuroprotective. To examine the role of more specific CDK inhibitors as potential neuroprotective agents, we studied the effects of roscovitine in TBI. Central administration of roscovitine 30 mins after injury resulted in significantly decreased lesion volume, as well as improved motor and cognitive recovery. Roscovitine attenuated neuronal death and inhibited activation of cell cycle pathways in neurons after TBI, as indicated by attenuated cyclin G1 accumulation and phosphorylation of retinoblastoma protein. Treatment also decreased microglial activation after TBI, as reflected by reductions in ED1, galectin-3, p22PHOX, and Iba-1 levels, and attenuated astrogliosis, as shown by decreased accumulation of glial fibrillary acidic protein. In primary cortical microglia and neuronal cultures, roscovitine and other selective CDK inhibitors attenuated neuronal cell death, as well as decreasing microglial activation and microglial-dependent neurotoxicity. These data support a multifactorial neuroprotective effect of cell cycle inhibition after TBI—likely related to inhibition of neuronal apoptosis, microglial-induced inflammation, and gliosis—and suggest that multiple CDKs are potentially involved in this process.
Introduction
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality in children and adults. In 2003, there were more than 1.5 million TBI-related injuries in the United States, resulting in approximately 1.2 million emergency room visits, 300,000 hospitalizations, and 50,000 deaths. Annually, about 230,000 TBI survivors experience long-term disability from cognitive, emotional, sensory, motor, and other impairments, making TBI a major public health problem.
In addition to causing direct mechanical injury, trauma initiates secondary cascades of biochemical and cellular changes, which substantially contribute to subsequent tissue damage and related neurologic deficits. Activation of specific signaling pathways in response to mechanical trauma causes delayed neuronal apoptosis as well as inflammation and reactive astrogliosis. Recent work has suggested a pathophysiological role for cell cycle pathways in neuronal apoptosis associated with both acute and chronic neurodegeneration. Postmitotic cells, such as neurons, do not engage in cell cycle progression once they differentiate; thus, cell cycle proteins are generally downregulated in these cells. However, mature neurons have the capacity to reenter the cell cycle, a process that results in apoptosis rather than neuronal proliferation (Nguyen et al, 2002). Abortive reentry into the cell cycle has been shown to contribute to neuronal apoptosis in vitro and in vivo. Activation of cell cycle proteins has been implicated in in vitro neuronal cell death caused by excitotoxic stress, ceramide, β-amyloid, KCl withdrawal, trophic factor deprivation, and DNA damage of various etiologies (Kruman et al, 2004). Cyclin-dependent kinases (CDKs) may also participate in neuronal apoptosis in the developing brain, as well as in neuronal degeneration in the adult brain after cerebral ischemia, Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (Nguyen et al, 2003). Inhibition of cell cycle reactivation provides neuroprotection both in vitro and in vivo (Wang et al, 2002). Previous studies in our laboratory have examined the role of cell cycle activation in the pathophysiology of experimental brain and spinal cord injury (SCI). Activation of the cell cycle occurs after TBI in both neurons and glial cells but with different downstream consequences: cellular proliferation/activation in astroglial and microglial cells versus apoptosis in neurons and oligodendro-glial cells (Di Giovanni et al, 2005). Central nervous system (CNS) injuries, such as trauma and ischemia, cause both proliferation of astroglia resulting in glial scar formation and microglial activation associated with the release of proinflammatory molecules that contribute to neurotoxicity (Byrnes et al, 2007). Although microglial activation may have protective actions in some experimental models, inhibition of brain inflammatory responses has proved to be neuroprotective in various models of neurodegeneration, as well as after neurotrauma (Byrnes et al, 2007).
Progression through the cell cycle is controlled by the interaction of numerous cell cycle molecules, including cyclins, CDKs, and CDK inhibitors (CDKIs). Activation of these pathways, which regulate cell division in mitotic cells, causes cell death in mature neurons. Increased levels of G1-phase cyclins D and E associated with neuronal apoptosis have been shown in both in vitro and in vivo models of excitotoxicity (Park et al, 2000). Moreover, upregulation of cyclins D, A, and B has been associated with toxin-induced hippocampal damage, whereas increased protein levels of cyclin D1 and CDK4 appear to play a critical role in excitotoxin-induced neuronal cell death (Ino and Chiba, 2001). Increased protein levels of cyclin D1 and CDK4 have been linked to the programmed cell death of motor neurons after spinal cord ischemia (Sakurai et al, 2000). In rat fluid percussion TBI, injury significantly increases the expression of many cell cycle-related genes, while downregulating those of endogenous cell cycle inhibitors (Cernak et al, 2005). Treatment with the powerful cell cycle inhibitor flavopiridol decreases TBI-induced lesion volume and improves behavioral outcomes after TBI (Cernak et al, 2005), suggesting that modulation of the cell cycle after injury might provide a mechanism for neuroprotection. Unfortunately, flavopiridol is a relatively nonspecific cell cycle inhibitor that also inhibits transcription, which complicates interpretation of its mechanism of action.
In the present studies, we examined the effects of postinjury administration of the more specific cell cycle inhibitor roscovitine in a well-characterized rat TBI model. In parallel, we also examined the effects of roscovitine on microglial activation and microglial-dependent neurotoxicity using in vitro models.
Materials and methods
All protocols involving the use of animals complied with the Guide for the Care and Use of Laboratory Animals published by NIH (DHEW publication NIH 85-23-2985), and were approved by the Georgetown University Animal Use Committee.
Reagents
Roscovitine was obtained from LC laboratories (Woburn, MA, USA); purvalanol A (no. 540500), CDK1 inhibitor (no. 217695), and CDK4 inhibitor (no. 219477) were from Calbiochem (Gibbstown, NJ, USA). Etoposide was from Tocris (Ellisville, MO, USA).
Induction of Injury
Fluid percussion-induced TBI was performed as previously described (Yakovlev et al, 1997) with some modifications. Briefly, male Sprague—Dawley rats (400 ± 25 g) were housed in a 12 h light/12 h dark cycle with free access to food and water. Rats were anesthetized with sodium pentobarbital (60 mg/kg intraperitoneal), followed by intubation, ventilation, and implantation of a catheter in the tail artery to measure blood pressure. Temperature was measured rectally and maintained at 36°C to 37°C throughout the procedure. A 5 mm craniotomy over the left parietal cortex was placed midway between lambda and bregma and 3.9 mm from the vertex, and a metal female leur loc disc was cemented to the site. The other end of the attachment was then fitted to the fluid percussion device. Force of impact was controlled through a computer connected to the device through PowerLab (AD Instruments, Colorado Springs, CO, USA) and recorded through the Chart4 Windows 4.2 software program (AD Instruments). All rats were subjected to moderate injury (2.7 to 2.9 atm). Thirty minutes after injury, roscovitine (300 nmol in 10 μL saline; this dose was chosen after pilot dose-response studies) or an equal volume of vehicle (saline) was injected intracerebroventricularly, contralateral to the injury site. Animals were killed at 24 h or 7 days after injury (n = 3 to 5 per group) for immunocytochemical/stereological studies, or at 21 days for histology, behavior, and volumetric measurements (n = 7 to 8 per group).
Neurologic Assessment
Motor and cognitive functions were assessed after injury using a composite neuroscore, as previously described (Cernak et al, 2004), and the Morris water maze (MWM) as previously described (Faden et al, 2003), respectively. Briefly, components of the composite neuroscore include measures of forelimb flexion (right and left), lateral pulsion (right and left), and the ability to maintain position on an angle board with animals positioned head up, head down, and right and left horizontal positions, each rated 0 to 5. For MWM, the rats were trained to locate a hidden, submerged platform using visual cues. The surface of the water was made opaque with the administration of white, nontoxic paint (Crayola) to the water. During training, the platform was maintained in a constant location in a single quadrant. The rat was placed in the water facing the wall at one of four randomly placed locations around the circular pool and the latency to find the hidden platform within 90 secs was recorded. Trials were conducted on days 14, 15, 16, and 17 after injury. To control for motor impairment, rats were tested to locate a visible, elevated platform after the last training trial.
Microglial Cultures
Microglial cells were obtained from postnatal day 2 Sprague—Dawley rat pups and cultured as described previously (Byrnes et al, 2006).
Microglial Proliferation and Activation
Microglia were stimulated with lipopolysaccharide (LPS, 1 μg/mL; Sigma, St Louis, MO, USA) with or without roscovitine (100 μmol/L) pretreatment (60mins). Micro-glial proliferation was then assessed using the MTS assay (Promega, Madison, WI, USA) according to the manufacturer's protocols. Microglial activation reflected by nitric oxide (NO) production was assayed using the Griess reagent assay (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions.
Microglia-Induced Neurotoxicity
Rat primary cortical neuronal cultures were derived from E18 Sprague—Dawley rat cortices (Taconic, Germantown, NY, USA) as previously described (Mukhin et al, 1998). At day 7 in vitro, media from microglia that had been stimulated with LPS with or without roscovitine pretreatment were transferred to the neuronal cultures. At 24 h after incubation with microglial-conditioned media, the LDH release assay (CytoTox 96 nonradioactive cytotoxicity assay kit; Promega) was used to assess neuronal cell death.
Etoposide-Induced Neuronal Cell Death
For etoposide-induced neuronal cell death, primary neurons were pretreated with various CDKIs (roscovitine: 25, 50, 100 μmol/L; purvanalol A: 25, 50 μmol/L; CDK1 inhibitor: 25, 50, 100 μmol/L; and CDK4 inhibitor: 25, 50 μmol/L) followed by exposure to etoposide (50 μmol/L). After 24 h, neuronal cell death was determined using the LDH assay as described.
Immunocytochemistry/Stereology
At 24 h and 7 days after injury, animals were killed and the brains harvested for histopathological analyses as described (Di Giovanni et al, 2005). The brains were frozen-sliced on a cryostat at a thickness of 20 μm, collected at a 1:5 ratio. Selected slides from animals at 24 h after injury were stained with fluoro-jade C (Chemicon, Temecula, CA, USA) to identify degenerating neurons following the manufacturer's instructions.
For immunocytochemistry, the sections were processed as previously described (Byrnes et al, 2007), using one or more antibodies recognizing the following targets: markers of cell cycle activation, including cyclin G1 (sc-7865, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and phospho S780 retinoblastoma (Rb) protein (ab47763, 1:100; Abcam, Cambridge, MA, USA); markers of micro-glial activation, such as CD68 (clone ED1, MCA341R, 1:100; Serotec, Raleigh, NC, USA), galectin-3 (ab2785, 6 μL/mL; Abcam), p22PHOX (sc-20781, 1:50; Santa Cruz Biotechnology); the neuronal marker NeuN protein (clone A60, MAB37 at 1:100; Chemicon, Billerica, MA); and the astroglia marker glial fibrillary acidic protein (GFAP, MAB360, 1:400; Chemicon). Fluorescence microscopy imaging was performed using Zeiss 510 meta confocal laser scanning microscope (LSM 510 META, Zeiss, Thornwood, NY, USA) as previously described (Byrnes et al, 2007). The images were taken using the × 10 objective at 1,024 times 1,024 resolution covering a field of view of 921.36 times 921.36 μm; all images in the same series, including sham, vehicle, and roscovitine, using the same antibodies, were acquired using the same instrument parameters. By using the multi-tile image acquisition capabilities of Zeiss LSM 510 META with a motorized stage, 16 images were taken of adjacent areas of the brain. These images were automatically spliced together to create a final image having a field of view of 3,685.45 times 3,685.45 μm at a resolution of 4,096 times 4,096 pixels. Modest variations in image brightness between adjacent panels of the composite image are intrinsic to this method.
Automatic cell counting in multi-tile fluorescent images obtained as described above was performed using the ImageJ software, version 1.4 (NIH). The indicated number of images, obtained from independent sections and including the channel of interest, were first converted to 8-bit grayscale using the Image > Type command. Next, the background was set as indicated using the Image > Adjust > Threshold command followed by cell analysis including cell counting using the Analyze > Analyze Particles command. All images in the same series, including vehicle and roscovitine, using the same antibodies, were processed using the same analysis parameters and the entire image was used for counting. The numbers shown in the graphs represent the average number of cells per image for each treatment. Unlike stereology, which offers a quantitative assessment of the total cell number throughout the entire lesion volume, this method provides a semiquantitative representation of cell number per representative field (the multi-tiled images presented) of three to five sections. Detailed instructions are available at http://www.rsbweb.nih.gov/ij/docs/menus/analyze.html.
Unbiased stereology, using the Stereologer 2000 program (SRC, Chester, MD, USA), was applied for quantitation after standard immunohistochemistry staining as previously described (Byrnes et al, 2007). The multilevel sampling design in the Stereologer 2000 software, based on the fractionator sampling method, was used to estimate the cell numbers of a particular phenotype in the injured hemisphere. For cyclin G1 cell counting purposes, the area of injury was outlined using a × 1.5 objective, and cells within each area were counted using a × 40 objective. For phospho-Rb cell counting purposes, the area immediately adjacent to the site of injury was outlined as described above.
Histology
At 21 days after injury, animals were decapitated and tissue processed as above. Slices were stained with Gill's hematoxylin, followed by counterstaining in 2.5% eosin and coverslipped. Lesion volume was measured based on the Cavalieri method of stereology and performed on tissue obtained 21 days after injury, as previously described (Byrnes et al, 2007). The lesion area, including both the cavity and surrounding damaged tissue, was outlined using the Stereologer 2000 program to obtain the final volume measurements.
Statistics
Continuous variables subjected to repeated measures over a period of time (MWM) were analyzed using repeated-measures analysis of variance followed by a one-way at each time point (P < 0.05). t–test and one-way analysis of variance were performed for stereologic analysis with significance set at P < 0.05. All continuous data are shown as mean ± s.e.m. Ordinal measurements (composite neuro-score) were evaluated using the Kruskal—Wallis test with individual Mann—Whitney U–tests corrected for multiple comparisons. Statistical significance was set at P < 0.05.
Results
Roscovitine Reduces Lesion Volume and Improves Behavioral Outcomes Following Traumatic Brain Injury
As shown in Figure 1, stereological analysis of lesion volume at 21 days after TBI indicated that treatment with roscovitine decreased brain lesion volume by 37% compared with vehicle-treated animals (t = 5.073, P < 0.001). The average vehicle-treated lesion volume was 20.6 mm and the average roscovitine-treated lesion volume was 13.0 mm3. Roscovitine treatment also significantly improved the composite neuroscores compared with vehicle-treated animals at 14 and 21 days after injury (Kruskal—Wallis 8.346, P < 0.02; Figure 2A). Individual Mann—Whitney U–tests, corrected for multiple measurements, revealed that vehicle-treated injured animals achieved significantly lower neuroscores compared with sham (P < 0.005), and that roscovitine improved motor outcome compared with vehicle at both 14 (P < 0.02) and 21 days (P < 0.001) after injury.
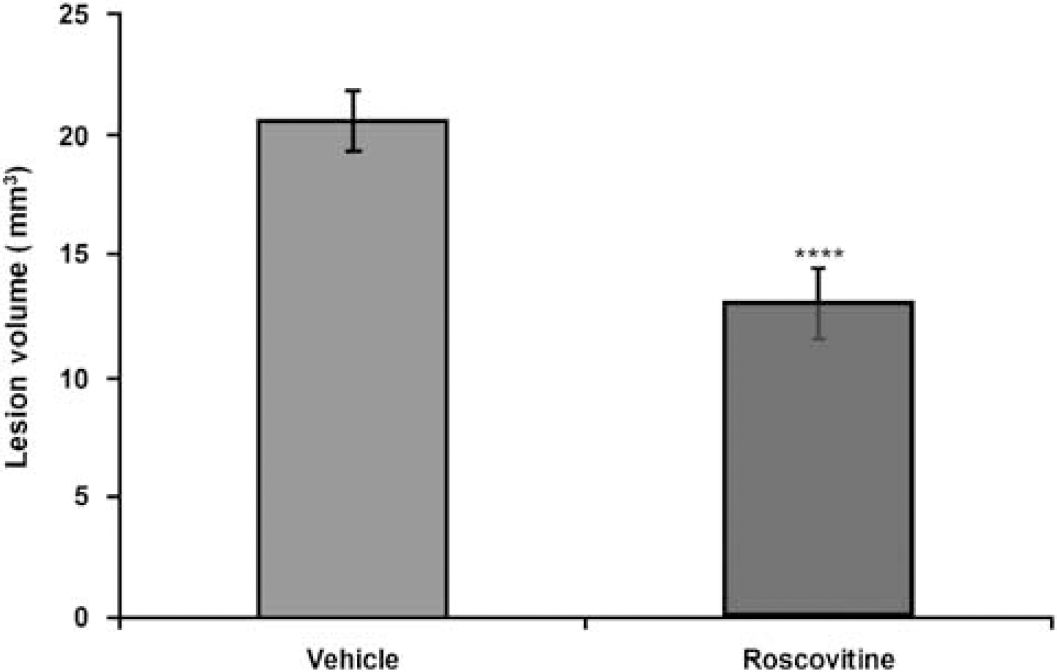
Treatment with roscovitine, 30 mins after TBI significantly decreases lesion volume. Lesion volume was measured 21 days after injury and compared with vehicle-treated animals (t = 5.073, ***P < 0.001; n = 7 to 8 per group)
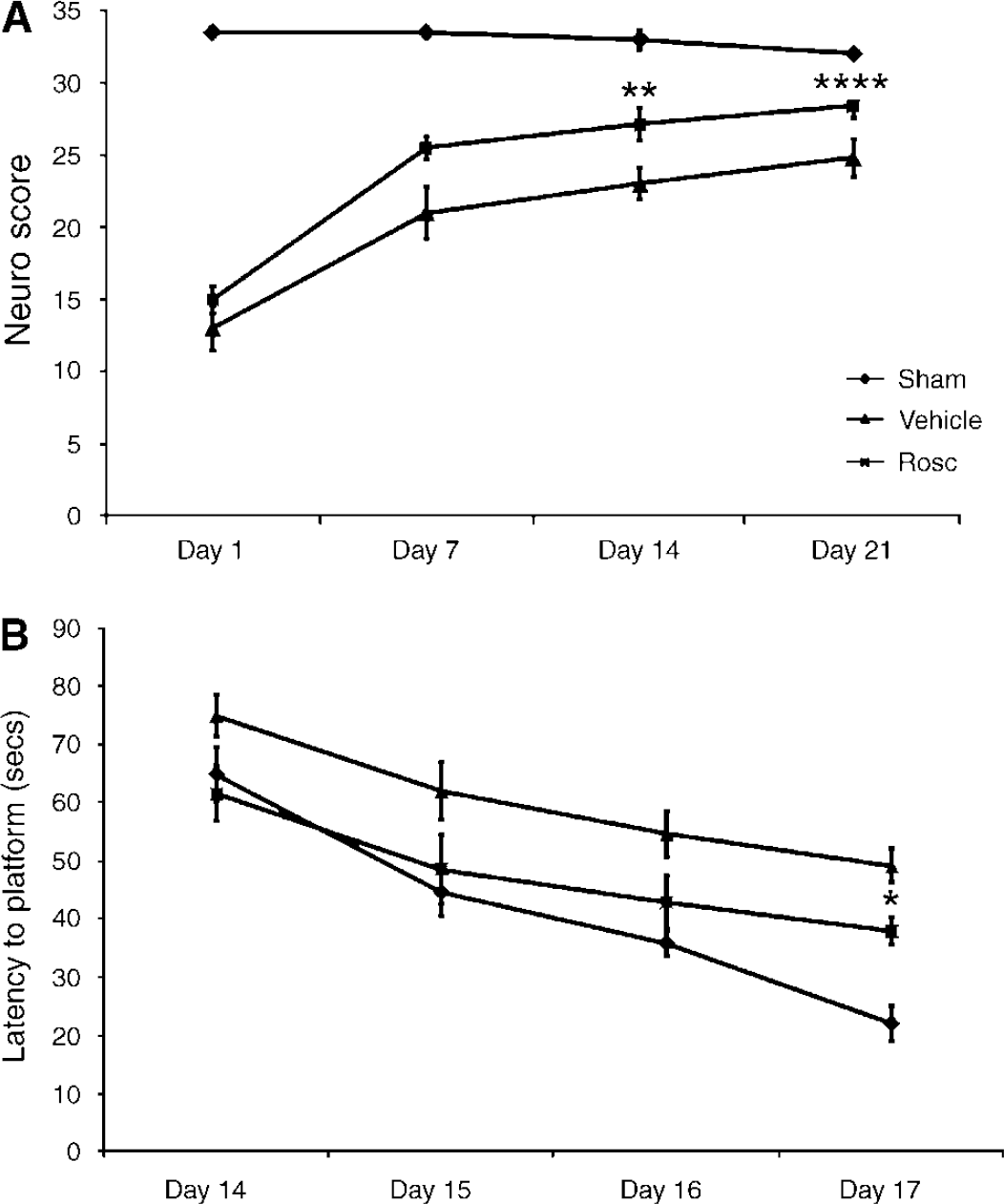
Administration of roscovitine (Rosc) significantly attenuates functional deficits induced by TBI. (
Analysis of cognitive function after injury showed a significant effect of time (F3,51 = 24.32, P < 0.0001) and treatment (F2,51 = 7.48, P < 0.005; Figure 2B). Latency to platform in the MWM test in vehicle-treated, injured rats was significantly longer on days 16 and 17 after injury (P < 0.05 and P < 0.001, respectively), when compared with sham-injured animals. However, there was no significant difference in performance between sham- and roscovitine-treated animals on day 16, suggesting that roscovitine induced improvement in cognitive function. By day 17 after injury, cognitive performance in roscovitine-treated injured animals was improved, when compared with vehicle-treated, injured animals (P < 0.05).
Roscovitine Decreases Neuronal Cell Death After Traumatic Brain Injury
To explore the effects of roscovitine on neuronal survival, we performed fluoro-jade C staining after TBI. Fluoro-jade C specifically stains dying neurons, which become fluorescent green. As shown in Figure 3A, tissue obtained 24 h after injury revealed fluoro-jade C-labeled degenerating neurons at the site of cortical injury lesion in vehicle-treated animals. Animals treated with roscovitine appeared to have a smaller injury lesion, which contained fewer fluoro-jade-C-positive cells. No significant fluoro-jade C staining was visible in sham animals (data not shown). We used the analytical capabilities of the ImageJ software to count fluoro-jade-positive cells in several independent sections, such as the one presented, from vehicle- and roscovitine-treated animals exposed to TBI; there is a significant decrease in fluoro-jade-positive cells in sections from roscovitine-treated animals (Figure 3B).
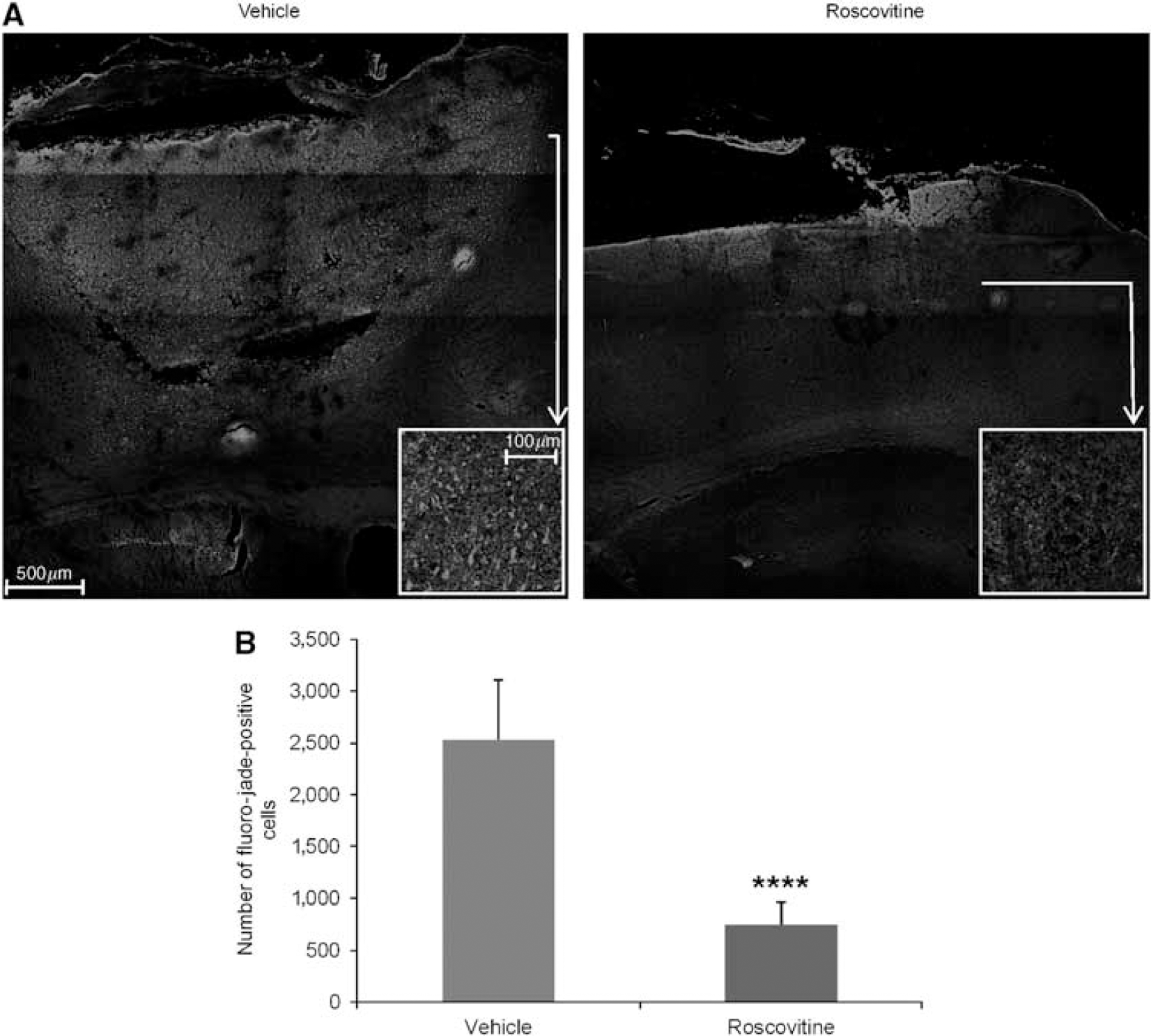
Roscovitine treatment decreases neuronal cell death as indicated by fluoro-jade C staining 24 h after injury. (
Roscovitine Significantly Attenuates the Numbers of Neurons Positive for Markers of Cell Cycle Activation
To explore the mechanisms responsible for roscovitine-induced neuroprotective effects, we examined two markers of cell cycle activation, cyclin G1 and phospho-Rb, which increase in apoptotic neurons following SCI, as we have previously shown (Di Giovanni et al, 2003). Staining with NeuN, a DNA-binding neuron-specific protein, was used to confirm the presence of cell cycle markers in neurons. Immunocytochemical analysis revealed that at 24h after injury, in vehicle-treated animals, there was an increased expression of cyclin G1 within the more central area of the lesion site (Figure 4A), whereas phospho-Rb was relatively more abundant on the periphery of the injury lesion (Figure 5A). Both markers colocalized with the NeuN immunostaining, indicating that cell cycle activation occurred in neurons. This elevated expression of cyclin G1 and Rb phosphorylation was reduced in tissue from animals treated with roscovitine (Figure 4A and Figure 5A), suggesting that the neuroprotective effect of this specific CDKI is associated with the downregulation of cell cycle pathways. We performed cell counting on several independent sections, such as the one presented, using ImageJ software; roscovitine treatment results in a significant decrease in the number of cyclin G1 and phospho-Rb, Figures 4B and Figure 5B, respectively. No significant cyclin G1 or phospho-Rb immunostaining was detected in sham animals (data not shown).
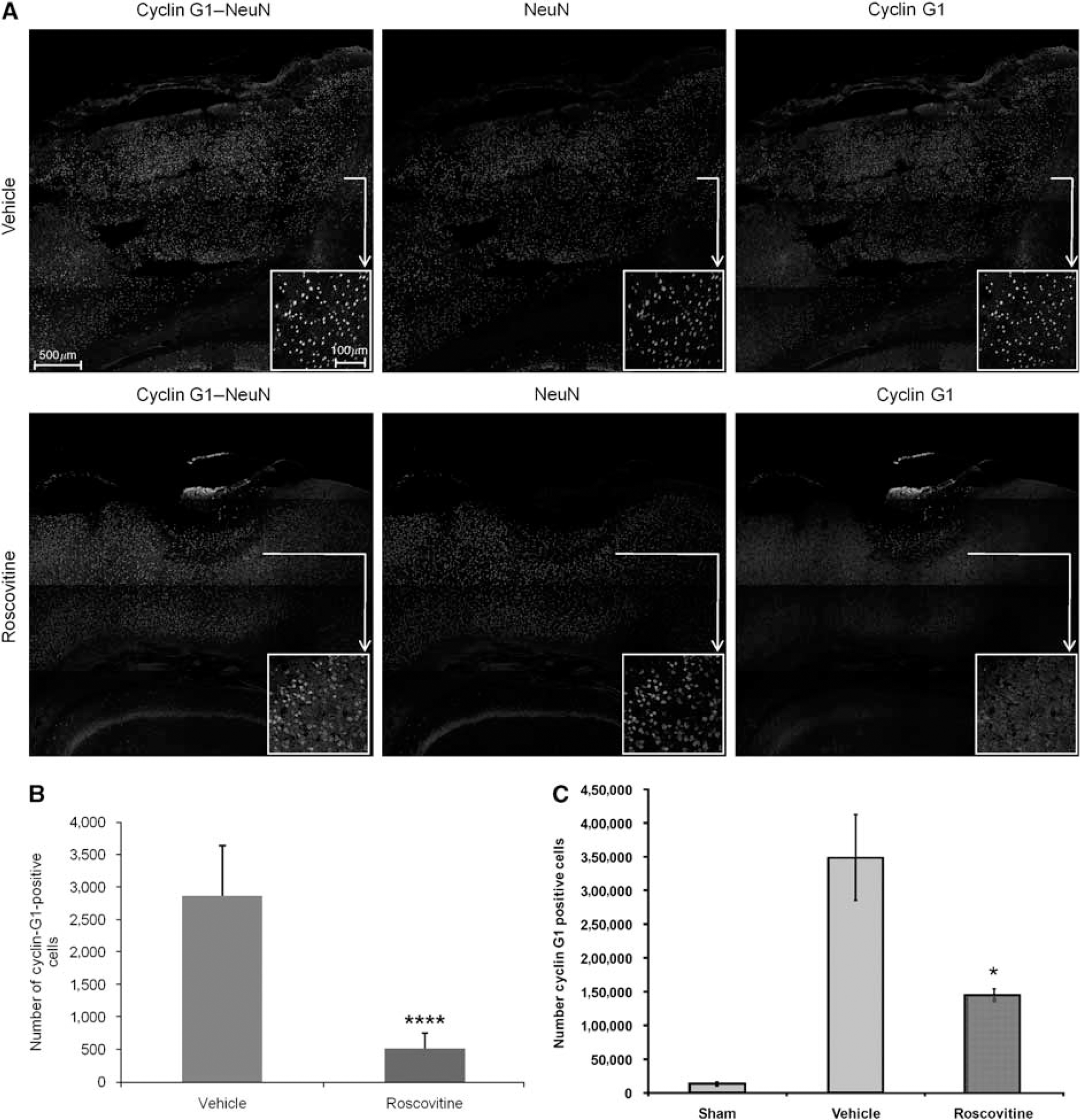
Roscovitine treatment significantly decreases expression of cyclin G1 in neurons 24 h after injury. (
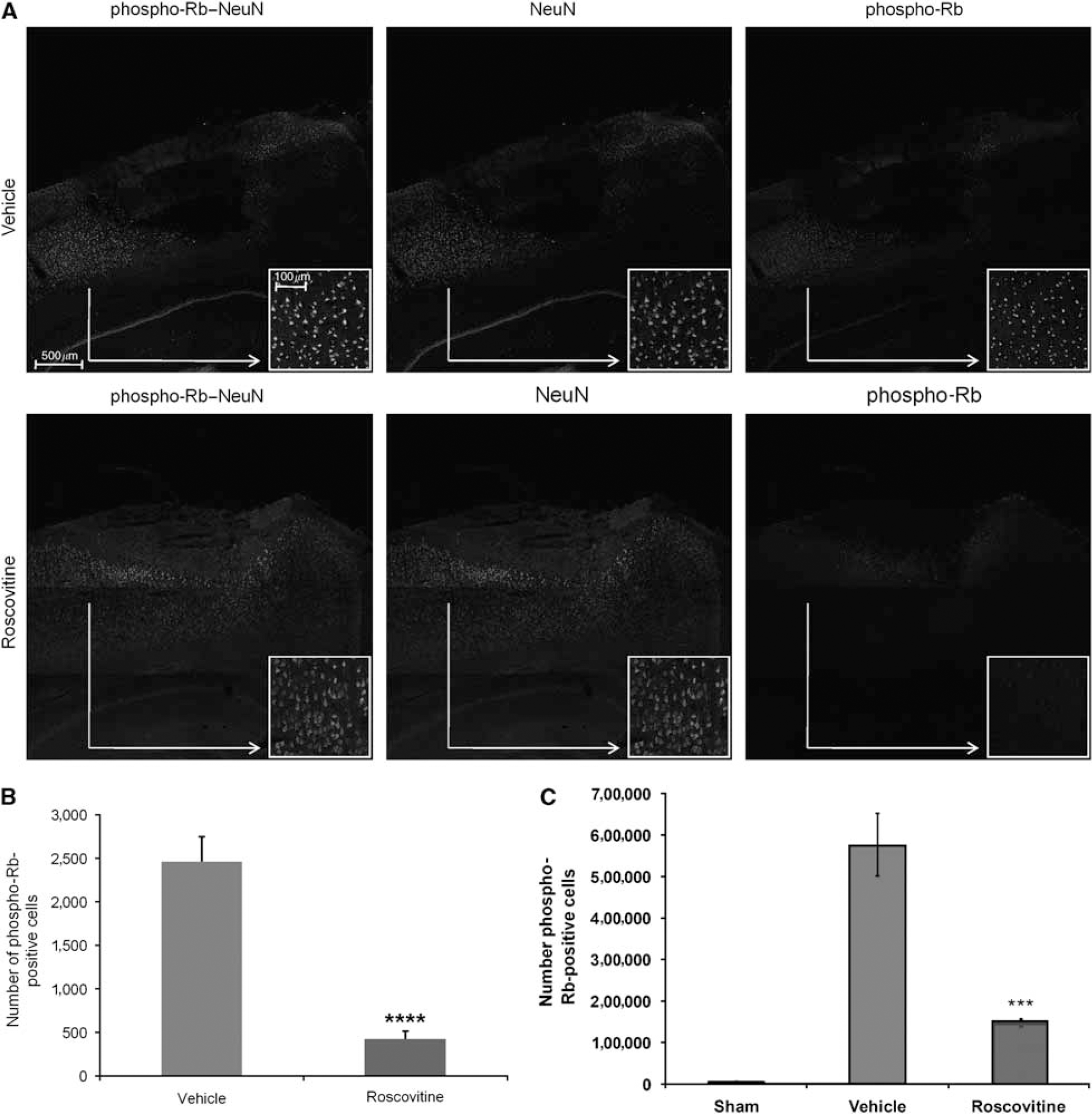
Roscovitine treatment significantly attenuates the phosphorylation of Rb in neurons 24 h after injury. (
To obtain a true quantitative assessment of the differences in cyclin G1 and phospho-Rb between vehicle- and roscovitine-treated animals, stereological analysis was performed at 24 h after injury. Injury induced a large increase in both cyclin G1 expression (F2,9 = 7.299, P < 0.02; Figure 4C) and phosphorylation of Rb (F2,10 = 27.386, P < 0.001; Figure 5C) compared with sham. Similar trends are observed with the two methods of cell counting, semiquantitative analysis of immunofluorescent images and quantitative stereology.
Roscovitine Decreases Activation of Astrocytes and Microglia After Traumatic Brain Injury
To examine the effects of TBI and cell cycle inhibition on astroglial and microglial activation, animals were treated with either vehicle or roscovitine after injury and killed 7 days later. In vehicle-treated animals, TBI increased the number of activated astroglia as evidenced by intensified staining for GFAP, an indicator of astrocyte reactive response to CNS injury (Brenner, 1994).
Injury also resulted in the activation of microglia as indicated by increased staining with four separate microglia markers—p22PHOX, galectin-3, ED1, and Iba-1. p22PHOX is a critical component of the microglial NADPH oxidase complex (DeLeo and Quinn, 1996), responsible for the generation of reactive oxygen species and involved in microglial; proliferation/activation (Byrnes et al, 2006). The activated microglia indicated by p22PHOX infiltrated the core of the lesion site, whereas activated astroglia appeared to surround the lesion site. (Figure 6A). In contrast, animals treated with roscovitine showed decreased expression of both GFAP-positive astroglia as well as p22PHOX-positive microglia (Figure 6A), suggesting that cell cycle inhibition attenuates both astrocyte activation and microglial activation associated with brain injury. Cell counting on several similar sections using the ImageJ program shows a significant decrease in the numbers of GFAP- and p22PHOX-positive cells after roscovitine treatment (Figure 6A).
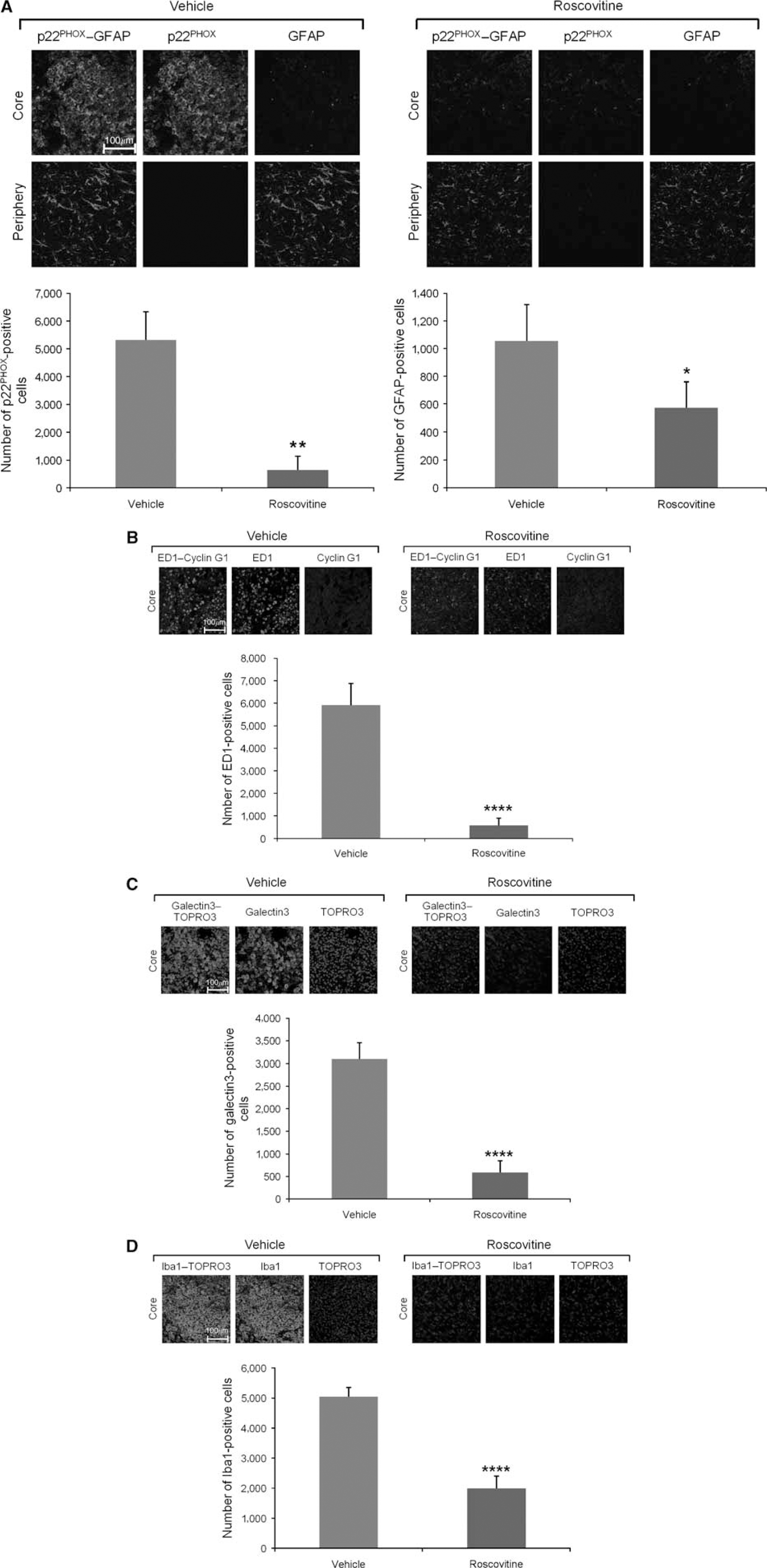
Roscovitine treatment decreases activation of microglia and astroglia at 7 days after injury, as indicated by immunostaining for p22PHOX, ED1, galectin-3, and GFAP All images are cropped from the indicated areas of larger multi-tile images (data not shown); multi-tile images are used for the cell counting data presented. (
In vehicle-treated animals, TBI induced an increase in the number of activated microglia, as evidenced by staining for ED1, a lysosomal membrane glycoprotein that is a marker of microglial phagocytic activation (Damoiseaux et al, 1994) (Figure 6B). The ED1-positive microglia are not costained with cyclin G1, suggesting that induction of this cell cycle protein is not involved in microglial activation. Animals treated with roscovitine showed decreased expression of ED1-positive microglia, suggesting that cell cycle inhibition attenuates microglial activation associated with injury. Cell counting on similar sections using the ImageJ program shows a significant decrease in the number of ED1-positive cells after roscovitine treatment (Figure 6B). The presence of activated micro-glia in the lesion site after TBI was also confirmed by immunostaining for galectin-3 (MAC-2 antigen, a β-galactoside-specific lectin), which is selectively expressed by activated microglia in the brain (Walther et al, 2000). In vehicle-treated animals, TBI increased the number of microglia with intense galectin-3 staining in the injury core (Figure 6C). Animals treated with roscovitine showed a marked decrease in galectin-3-positive microglia, suggesting that cell cycle inhibition attenuates microglial activation associated with brain injury. Cell counting on similar sections using the ImageJ program shows a significant decrease in the numbers of galectin-3-positive cells after roscovitine treatment (Figure 6C). Finally, microglial activation was also confirmed by immunostaining for Iba-1, which is a calcium-binding protein specifically expressed in brain microglia and upregulated on microglial activation (Ito et al, 2001). In vehicle-treated animals, TBI increased the number of microglia with strong Iba-1 staining in the injury core (Figure 6D). Animals treated with roscovitine showed a marked decrease in Iba-1-positive microglia, further confirming that cell cycle inhibition attenuates micro-glial activation associated with brain injury. Cell counting on similar sections using the ImageJ program showed a significant decrease in the numbers of Iba-1-positive cells after roscovitine treatment (Figure 6D). No significant cyclin G1, galectin-3, p22PHOX, or GFAP immunostaining was detected in sham animals (data not shown).
Roscovitine Decreases Activation of Primary Microglia in In Vitro Culture Models
To further investigate the potential of roscovitine to attenuate microglial activation, we used an in vitro culture system based on primary rat brain microglia stimulated by LPS. Pretreatment with roscovitine significantly attenuated markers of microglial acti-vation such as proliferation (Figure 7A) and release of NO, a well-known neurotoxic agent (Figure 7B). In this model, we also examined other CDKIs with relatively narrow specificities, including a CDK1-selective inhibitor and a CDK4-selective inhibitor. The CDK1 inhibitor is approximately 5 times more selective for CDK1 compared with CDK5 (Andreani et al, 2000). The CDK4 inhibitor is 500 times more selective for CDK4 compared with CDK2 (Kubo et al, 1999). Pretreatment with both CDK1 and CDK4 inhibitors significantly attenuated both microglial proliferation (Figure 7C) and release of NO (Figure 7D).
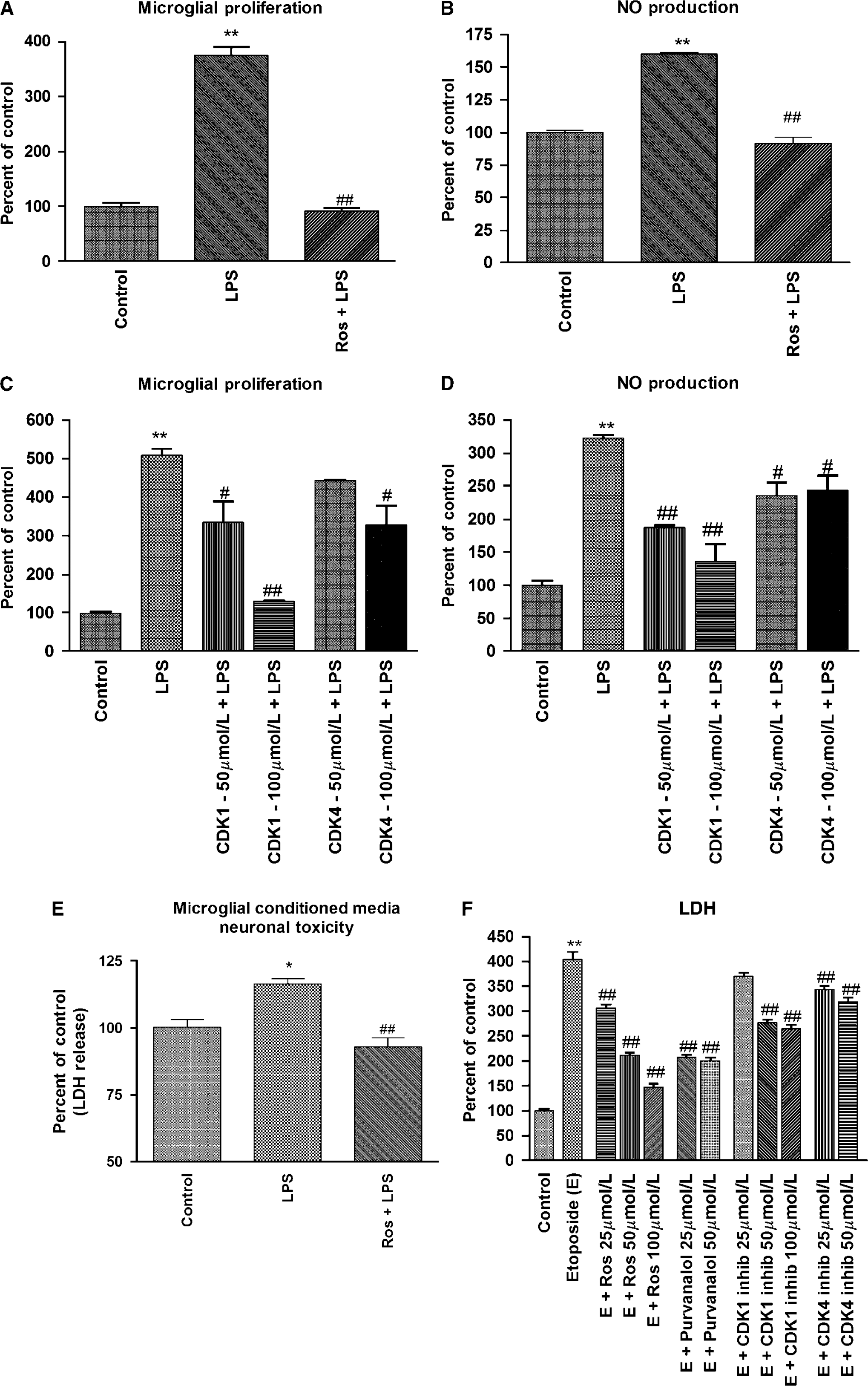
Roscovitine reduces activation of primary microglia and primary neuronal cell death in in vitro culture models. Primary rat microglia were incubated with LPS (1 μg/mL) for 24 h and activation was determined by proliferation assays (
We used an in vitro model of mixed primary brain microglia-neuronal cultures to evaluate the potential of roscovitine to attenuate microglia-dependent neurotoxicity. Pretreatment of microglia with roscovitine before LPS treatment for 24 h significantly attenuated the ability of microglial conditioned media to induce neuronal cell death (Figure 7E). These data suggest that roscovitine decreases the release of neurotoxic compounds by activated microglia.
Finally, we examined the effects of roscovitine as well as other CDKIs in a well-established model of neuronal apoptosis induced by etoposide. In addition to roscovitine, CDK1 and CDK4 inhibitors, we also used purvalanol A, a compound that has a CDK inhibitory profile similar to roscovitine and may be used to confirm roscovitine-based data indicating the involvement of CDKs in a process (Bain et al, 2007). As shown in Figure 7F, all the tested CDKIs were able to significantly attenuate etoposide-induced neuronal cell death, suggesting the involvement of multiple CDKs in neuronal apoptosis.
Discussion
Over the past several years, our group has studied the role of cell cycle activation in the pathophysiology of CNS trauma. After SCI (Byrnes and Faden, 2007; Di Giovanni et al, 2003) or TBI (Cernak et al, 2005) in rats, increased expression of cell cycle proteins occurred in neurons showing caspase-3 activation and morphologic signs of apoptosis. Cell cycle activation after TBI was also associated with astrogliosis and microglial activation (Di Giovanni et al, 2005). Treatment with flavopiridol reduced behavioral and histologic abnormalities after TBI (Di Giovanni et al, 2005) and SCI (Byrnes et al, 2007). Protective effects of flavopiridol have also been reported in cerebral ischemia (Wang et al, 2002).
Flavopiridol, a semisynthetic flavonoid, is a potent but nonselective competitive CDKI acting on all CDKs examined thus far (Newcomb, 2004). Unfortunately, it also potently inhibits the positive transcription elongation factor b (of which CDK9 is a component) and consequently acts as a global suppressor of transcription (Monaco et al, 2004). In contrast, roscovitine is a purine analog that competitively inhibits CDKs in a more selective fashion, acting preferentially on CDKs 1, 2, and 5, and possibly CDKs 7 and 9 (Meijer et al, 1997). It poorly inhibits CDKs 4 and 6 (Meijer et al, 1997) and does not significantly inhibit members of most other kinase families (Bain et al, 2003). Overall, multiple studies have confirmed that roscovitine is among the most potent and specific CDKIs (Bach et al, 2005; Bain et al, 2007; Meijer et al, 1997). Moreover, although roscovitine can inhibit CDK9 in vitro,it does not appear to interact with it in vivo; this may explain why, unlike flavopiridol, roscovitine does not globally inhibit gene expression (Lam et al, 2001). Because both RNA and protein synthesis may be required for induction of neuronal apoptosis, it is likely that the ability of flavopiridol to attenuate neuronal cell death partly reflects its transcriptional repressor role. Therefore, to better address whether activation of CDKs/cell cycle activation is an important mechanism for TBI-induced cellular changes and related functional deficits, we examined the effect of roscovitine treatment.
Central administration of a single dose of roscovitine 30 mins after injury markedly decreased lesion volume and significantly improved behavioral out-comes, when compared with vehicle-treated animals. Quantitative stereological techniques and high-resolution confocal microscopy were used to characterize the molecular and cellular changes induced by TBI and its modulation by roscovitine. After TBI, activation of neuronal cell cycle pathways—including phosphorylation of Rb protein and increased expression of cyclin G1—as well as neuronal cell death occurs within 24 h; considerable microglial and astrocyte activation/proliferation were detected by 7 days. Roscovitine treatment significantly attenuated each of these changes. At 24 h, roscovitine-treated animals showed decreased numbers of neurons positive for phospho-Rb and cyclin G1 as compared with controls, and fewer degenerating neurons indicated by decreased fluoro-jade C staining. At 7 days after injury, roscovitine treatment reduced microglial activation, as shown by reduced numbers of cells positive for ED1, galectin-3, and p22PHOX, and decreased astrocyte activation, as indicated by fewer GFAP-positive cells. Decreased numbers of cyclin G1-positive cells were also found at this later time point, suggesting that roscovitine-induced inhibition of cell cycle activation after a single early injection is sustained.
The mechanisms by which the active cyclin/CDK complexes induce neuronal apoptosis are being clarified. One of the signal transduction pathways involved includes cyclin D-dependent CDK4/6, which phosphorylates Rb on specific residues. Phosphorylation of Rb causes dissociation of Rb from the Rb/E2Fs complex, with activation of E2Fs transcription factors; E2Fs induce apoptosis by upregulating cell death mechanisms, such as activation of B- and C-myb genes; by increasing expression of caspases 3, 9, and 8, and Apaf-1; and by activation of p53 and p73. These activities lead to increased expression of proapoptotic Bcl-2 family members and release of mitochondrial proapoptotic proteins (Nguyen et al, 2003). Recent reports indicate that in some models of neuronal cell death, activation of CDK5 is upstream of CDK4/6 and that CDK5 can directly phosphorylate Rb and initiate neuronal cell death (Hamdane et al, 2005). CDK5 is implicated in neurodegeneration and cell death; knockout of CDK5 results in neuroprotection after focal brain ischemia (Rashidian et al, 2005). CDK2 can also phosphorylate Rb (Lundberg and Weinberg, 1998). As roscovitine has no activity against CDKs 4 and 6, but is highly potent against CDKs 2 and 5 (Meijer and Raymond, 2003), our data suggest that activation of CDK2 and/or CDK5 may contribute to neuronal cell death after TBI, and that roscovitine might downregulate Rb phosphorylation by inhibiting CDK2/5. It is important to underscore that specificity limitations common to pharmacological interventions, such as the one used in our study, do not allow determination of the precise CDK that is targeted by roscovitine and is responsible for the protective effects observed. Furthermore, our data do not exclude the possibility that other CDKs besides CDK2 or CDK5 are involved in neuronal cell death or other cellular processes that are active in our model. As discussed above, multiple CDKs are often involved in sequential and/or parallel activation cascades when modulating cellular activity. Therefore, treatment effects may result from interventions at multiple points in the reaction cascade. Our in vitro data are consistent with this conclusion, showing that in addition to roscovitine and purvalanol A, which target multiple CDKs, etoposide-induced neuronal cell death and/or primary microglial activation in culture are also attenuated by somewhat selective CDK1 and CDK4 inhibitors. In addition, published reports indicate that significant redundancy exists in CDK signaling. For example, CDK2 knockout mice are viable and show no significant proliferation effects, despite previous data indicating an important function for CDK2 in the cell cycle; this suggests that other CDKs may be able to compensate for the loss of CDK2 (Berthet et al, 2003). Thus, targeting multiple CDKs may provide better neuroprotection than more selective inhibitors. A role for increased cyclin G1 expression and Rb phosphorylation in neuronal cell death has also been suggested in other experimental models, including ischemia, excitotoxicity, and neurodegeneration (Sultana and Butterfield, 2007). Cyclin G1 immunoreactivity and mRNA are low in a mature rodent brain; however, after injury, their expression increases in the nuclei of damaged neurons. Cyclin-G1-positive neurons also correlate strongly with degenerating neurons in a model of permanent middle cerebral artery occlusion (Maeda et al, 2005).
Phosphorylated Rb induced by hypoxia-ischemia and middle cerebral artery occlusion is expressed in pyknotic cells and neurons both in the injury core and peri-infarct regions (Rashidian et al, 2005). In our model, cyclin G1 was expressed preferentially, although not exclusively, in neurons in the central area of the lesion, whereas phospho-Rb was expressed relatively more in the neurons toward the periphery of the injury lesion. This might suggest differential activation of cell cycle components in the central lesion area versus penumbra, although variations in the time course of cell cycle activation may provide an alternative explanation.
Cyclin G1 overexpression induces cell death (Okamoto et al, 1999), although the mechanisms responsible for cyclin G1 induction after injury and related neuronal cell death are not well characterized. Some recent data suggest that cyclin G1 is a target gene of the p53 tumor suppressor protein, a well-known inducer of apoptotic cell death. In this case, cyclin G1 would be downstream of the signaling pathway involving phosphorylation of Rb and activation of E2Fs transcription factors that lead to p53 activation. Other data indicate that cyclin G1 is associated with CDK5 and plays a role in G2/M transition (Seo et al, 2006); under these conditions, increased activation of cyclin G1 may induce apoptosis of mature neurons that are unable to divide.
Although CNS injury-induced aberrant cell cycle activation induces apoptosis in postmitotic cells, it initiates proliferation in mitotic CNS resident cells, such as astrocytes and microglia (Takuma et al, 2004). The functional significance of microglial proliferation and activation has been debated. On the one hand, activated microglia produce various proinflammatory molecules, such as IL-1/β, NO, complement components, MCP-1, osteopontin, MHC classes I and II, and reactive oxygen species (produced by the NADPH oxidase enzyme), and these have been implicated in secondary injury (Cernak et al, 2005). On the other hand, microglial-related factors, such as TNFa, may participate in both secondary injury and recovery (Tamatani et al, 1999), whereas others, such as glial cell line-derived neurotrophic factor, may provide endogenous neuroprotection (Hashimoto et al, 2005). Similarly, although astrocytes play an important role in maintaining normal brain function, changes after injury, such as swelling and/or hypertrophy (astrogliosis) as well as proliferation (astrocytosis), may serve pathophysiological actions (Cernak et al, 2005). For example, the glial scar associated with astrocytosis may inhibit regeneration and plasticity after brain injury (Silver and Miller, 2004). A recent in vitro study of ischemia has suggested that the neurotoxic versus neuroprotective actions of activated microglia may in part reflect injury severity (Lai and Todd, 2008).
We have shown previously that cell cycle proteins are upregulated in microglia after SCI as well as in primary microglia in vitro (Byrnes et al, 2006), and that activation of astrocytes and microglia occur after SCI and are correlated with cell cycle initiation (Byrnes et al, 2007). Another group has reported cell cycle activation in microglia after SCI and protective actions by the cell cycle inhibitor olomoucine (Tian et al, 2007). We established with a high degree of confidence the postinjury microglia status in the brain by using four independent markers of micro-glial activation: ED1, galectin-3, p22PHOX, and Iba-1 (Byrnes et al, 2006; Ito et al, 2001). The present studies show that microglial activation occurs after TBI, as indicated by the greatly increased number of cells positive for the three markers. These changes are attenuated by roscovitine. Roscovitine treatment also inhibited TBI-induced activation of astrocytes, as shown by decreased staining with the GFAP marker of astrocyte reactive response (Brenner, 1994). Because the complex cellular environment in the brain makes it difficult to determine with precision the primary action of any pharmacological agent, we used cell culture models of primary microglia and neurons to analyze the effects of roscovitine on specific cell types. In previous in vitro studies, we have shown that roscovitine is protective against neuronal cell death resulting from etoposide-induced DNA damage, and that roscovitine inhibits astrocyte proliferation (Cernak et al, 2005). In the present study, we focused on the effects of roscovitine on primary microglia using a well-established model of LPS-induced activation. In this model, roscovitine attenuated microglial proliferation and the release of neurotoxic mole-cules such as NO induced by LPS. Furthermore, using a model of mixed primary microglia/neuronal cultures, we show that roscovitine markedly re-duced microglial-dependent neurotoxicity. CDK1 and CDK4 inhibitors also attenuate microglial activation in our in vitro model, suggesting the complexity of the CDK cascade that regulates microglial functions.
In conclusion, roscovitine treatment after TBI markedly reduces lesion volume and improves behavioral outcome. Treatment attenuates cell cycle activation in neurons, astrocytes, and microglia; effects are associated with reductions in neuronal apoptosis, glial scar formation, and microglial activation. These diverse effects of roscovitine are supported by complementary cell culture observations, and such pluripotential actions may explain its robust neuroprotective activity.