
Abstract
In the present study, we observed the expression of toll-like receptor 4 (TLR4) and its downstream signal pathway in peripheral blood monocytes (PBMs) from patients with acute cerebral infarct (ACI). The expression of TLR4 and MyD88 by PBMs was determined by flow cytometry and reverse transcriptase-polymerase chain reaction, and nuclear factor-κB (NF-κB) activity was detected by electrophoretic mobility shift assay. Ischemia/reperfusion injury-induced cerebral edema, infarction area, and neurologic impairment scores were determined in MyD88 gene knockout mice. The results indicated a significant increase in circulating TLR4+ monocytes in ACI patients as compared with the control group and the transient ischemia attack (TIA) group. This change paralleled an elevation in TLR4mRNA transcription and serum tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 in the ACI and TIA groups. Correlation analysis showed TLR4 expression to significantly correlate with cytokine levels and stroke severity. MyD88mRNA differed insignificantly among the three groups. Compared with wild-type mice, 6 h of cerebral ischemia followed by 24 h of reperfusion did not significantly change cerebral edema, cerebral infarction area, and neurologic impairment scores in MyD88 gene knockout mice. Compared with the control group, serum heat shock protein (HSP) 60 increased significantly in the ACI and TIA groups, leading to NF-κB activation in TLR4/CD14-transfected HEK293 cells. It is suggested that upregulated TLR4 expression on PMBs may act as one of the peripheral mechanisms of inflammatory injury after ACI. Moreover, circulating HSP60 may be a ligand for TLR4, which is involved in the peripheral mechanism of inflammatory injury after ACI, possibly through an MyD88-independent signal pathway.
Introduction
Cerebral infarction, the most common acute cerebrovascular disease with a high attack rate, death rate, and disability rate, is one of the three leading causes of human deaths. The pathogenesis of this disease has not been elucidated yet. Ischemic/hypoxic injury of brain tissues and subsequent necrosis and apoptosis of nerve cells had long been considered the principal pathophysiological mechanism of cerebral infarction (Mergenthaler et al, 2004; Frizzell, 2005; Mogi et al, 2006). In recent years, however, the role of inflammatory reaction in brain injury following cerebral infarction has been recognized. Several inflammatory events have been identified after cerebral ischemia, including aggregation of inflammatory cells, and upregulation of cytokines and intercellular adhesion molecules. Antiinflammatory therapy has become conspicuous for its neuroprotective effect in animal models, suggesting inflammatory reactions to be key to ischemia/reperfusion injury of brain tissues (Kim et al, 2005; Pan et al, 2007). Nevertheless, the mechanism involved is left to be elucidated.
Toll-like receptors (TLR) are members of an evolutionarily conserved family, which mediate innate immunity. There are many TLR4 ligands (Poltorak et al, 1998), primarily lipopolysaccharide (LPS) and others, such as bacterial muramic acid, flagella, high-mobility group box-1, and heat shock proteins (HSPs) (Triantafilou and Triantafilou, 2004; Tsan and Gao, 2004). When bound to its ligands, TLR4 activates nuclear factor-κb (NF-κB) through MyD88 and induces high production of cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-6, and finally leads to intense inflammation. Hence, TLR4 plays a key role in sepsis and septic shock (Miyake, 2004). Moreover, TLR4 was shown to play a key role in atherogenesis (Michelsen et al, 2004), inflammatory injury of the heart (Oyama et al, 2004), and ischemia/reperfusion injury of the liver (Tsung et al, 2005) and brain (Cao et al, 2007; Caso et al, 2007). Acute cerebral infarct (ACI) may result in intense peripheral inflammatory reaction (Offner et al, 2006), inflammatory injury of the blood—brain barrier, and aggregation of peripheral cells in the brain. Therefore, we hypothesize that TLR4 takes part in inflammatory injury after cerebral infarction, possibly through a peripheral mechanism. To test the hypothesis, the expression of TLR4 and MyD88 in peripheral blood monocytes (PBMs), that is, lymphocytes and monocytes, was determined in this study; meanwhile, the changes in the downstream signal molecules TNF-α and IL-6 after TLR4 activation were analyzed. In addition, the possible ligands for peripheral TLR4 were examined.
Subjects and methods
Study Subjects
Study subjects were selected according to the inclusion and exclusion criteria adopted by Garlichs and Goldstein (Garlichs et al, 2003; Goldstein et al, 2004). A total of 49 ACI patients and 16 transient ischemia attack (TIA) patients who were admitted to the Stroke Unit of the department within 24 h after the onset of the symptoms were selected. All the patients were diagnosed and classified according to the criteria adopted by the Trial of Org 10172 in Acute Stroke Treatment (TOAST) (Kolominsky-Rabas et al, 2001). The exclusion criteria included intracerebral hemorrhage (confirmed by computed tomography or magnetic resonance imaging); significant stenosis of extra- and intracerebral carotid arteries (confirmed by TCD and ultrasonography); arrhythmias and intracardiac thrombi (confirmed by electrocardiography and echocardiography); symptoms lasting more than 24 h; obvious inflammatory conditions (e.g., infectious disease, systemic lupus erythematosus, rheumatism, and rheumatoid disease) within the half year before enrolment; hospital-acquired infection (confirmed by the patient's symptoms and laboratory findings); acute myocardial infarction (diagnosed according to the patient's symptoms and electrocardiographic findings); acute ischemia of liver (confirmed by liver function test); autoimmune disease (diagnosed according to the patient's symptoms and positive autoimmune antibodies); patients who took glucocorticosteroids or immunodepressants; patients who were incompliant to the study protocol or could not undergo all the tests required by the study. A total of 25 healthy people who underwent regular physical examinations at our hospital were included in the control group. The controls were matched with the patients in terms of age and gender, and they did not have acute ischemia of the heart, peripheral tissues, or brain within 12 months before enrolment (see Table 1). Signed informed consent was obtained from all the study subjects and the study protocol was approved by the Medical Ethics Committee of the hospital. Peripheral leukocytes and platelets were counted, and the concentration of C-reactive protein was measured.
Baseline data of subjects
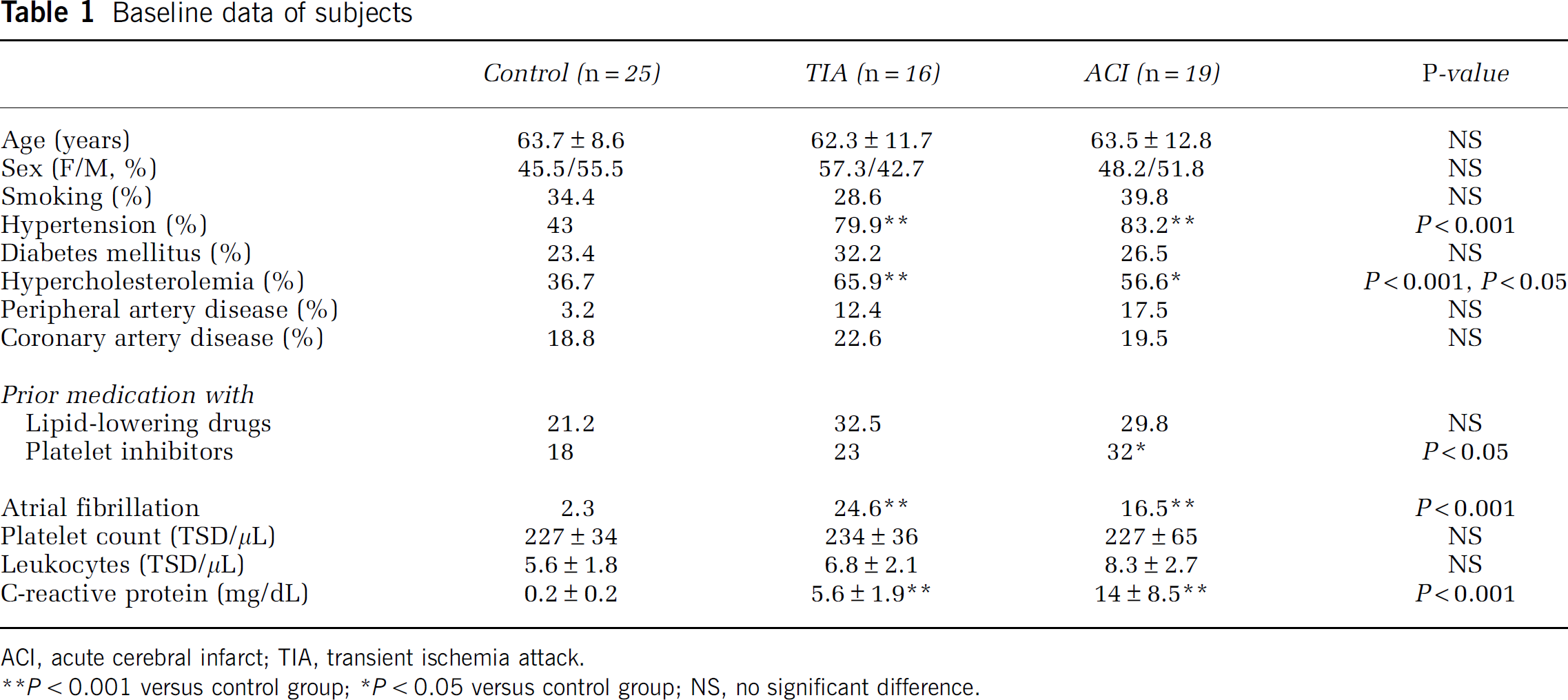
ACI, acute cerebral infarct; TIA, transient ischemia attack. ACI, acute cerebral infarct; TIA, transient ischemia attack.
P < 0.001 versus control group;
P < 0.05 versus control group; NS, no significant difference.
Among the ACI patients with the onset of symptoms within 3 h before admission, thrombolysis was administered only in two patients according to the stroke criteria of the National Institute of Neurological Disorders and the AHA guidelines (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995).
The National Institutes of Healthy Stroke Scale (NIHSS) was used to rate the severity of stroke (Lyden et al, 1994), and modified Rankin scores (mRS) were used to assess the functional status of patients at 3 months after the onset of stroke.
Sample Collection and Preparation
A total of 11 mL of blood sample was drawn from the median cubital vein of one patient after he had stayed in the hospital for 18 h and in the other patients immediately after admission; 9 mL of the blood sample was mixed thoroughly with heparin (50 U/μL blood). Of the anti-coagulated blood, 8 mL was treated with Ficoll solution (Shanghai Chemistry Agents Factory) to isolate monocytes, and the other 1 mL was subjected to flow cytometry. The 2 mL not treated with heparin was used to isolate serum at room temperature, and the serum obtained was frozen and stored at −20°C for later use.
Determination of TLR4 Expression by Flow Cytometry
As described previously (Geng et al, 2005), phycoerythrin-crosslinked rat anti-human TLR4 monoclonal antibody (eBioscience, San Diego, CA, USA) was used for flow cytometry, and phycoerythrin-cross-linked rat IgG2a antibody was used as the negative control. Blood cells were divided into mononuclear cells, lymphocytes, and neutrophils according to the flow cytometric parameters FS and SS. Then TLR4 expression was detected in CD14 + mononuclear cells that were labeled by anti-CD14 monoclonal antibody (eBioscience). A total of 20 μL phycoerythrin-crosslinked rat anti-human TLR4 was incubated for 30 mins with 100 μL whole blood at room temperature in dark, mixed with a certain volume of the akaryocyte lysis buffer, and then washed twice with sterile water containing bovine serum albumin and sodium azide. Lymphocytes, monocytes, and neutrophils were precipitated with 500 μL sterile water, and then TLR4 expression by the cells was detected by flow cytometry (BeckmanCoult, Fellerton, CA, USA).
Detection of TLR4 and MyD88 mRNA by RT-PCR
Total RNA was extracted from PBMs using one-step Trizol kit (Gibco Co., Carlsbad, CA, USA). Each sample was mixed with the same amount of reverse transcriptase to synthesize cDNA using Bio-Rad Cycler according to the kit manufacturer's instructions. Glyceraldehyde 3-phosphate dehydrogenase was used as the internal standard. The primers were synthesized by Shanghai Sangon Biological Engineering Ltd Co. (Shanghai, China). The optimal primer sequences, annealing temperatures, and amplification cycle numbers were determined by a series of pilot experiments (Table 2).
Reverse transcriptase-PCR primer sequences, sequence accession numbers, annealing temperatures, and cycle numbers

GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
The samples were analyzed in duplicate, and subjected to agarose gel electrophoresis and incubated at room temperature with molecular probes for 30 mins. Fluorescence signal was observed by UV transillumination at 300 nm. The samples were quantitatively analyzed for fluorescence density using BioRad Fluor-S MultiImager and Quantity One Software. The transcript levels were corrected by dividing the values of the amplified products by the mean value of glyceraldehyde 3-phosphate dehydrogenase.
Construction of Mouse Cerebral Ischemia—Reperfusion Models
Male MyD88-knockout mice (8 weeks old, weighing 18 to 22 g) were purchased from American Jackson Laboratories (Bar Harbor, ME, USA) and bred with C57BL/c mice (wild-type mice) in our laboratory. All mice were kept and bred in-house under pathogen-free condition. The Animal Care and Use Committee of the Third Military Medical University approved the experimental protocol. We constructed the mouse models of middle cerebral artery reperfusion, as described previously (Cao et al, 2007). The mice were anesthetized by intraperitoneal injection of 2% pentobarbitol sodium (40 mg/kg), immobilized in a dorsal position, and incised at the cervical median line, followed by isolation of the left common carotid artery (CCA), external carotid artery, internal carotid artery, and the deep branch pterygopalatine artery. After that, the CCA was ligated at the proximal part and the external carotid artery at the distal part, leaving the ligature suture at the proximal part of the CCA crotch. After occlusion of the pterygopalatine artery and the distal internal carotid artery with mini-bulldog clamps, we incised at the site (1.0 mm away from the CCA crotch) with microscissors, and inserted prepared nylon silk into the internal carotid artery until it reached the cerebral prerolandic artery (about 8.29 ± 0.54 mm depth) and blocked the blood supply of the middle cerebral artery. Thereafter, we tightened the ligature line at CCA, and performed regular saturation on cervical incision. Then, we gently withdrew the nylon silk to the CCA crotch, maintained the temperature at about 25°C during and after surgery, and exposed the animals to incandescent lamp to keep their rectal temperature at (37 ± 1)°C until its palinesthesia. The signs of a successful model are (1) left Horner symptom and (2) right-side hemiparalysis mainly for the forward limb.
Neurologic Impairment Scores
Two hours after the surgery, the mice awoke from narcosis. We then screened the model according to the standard of Longa 5 (Longa et al, 1989) grading method, as described previously (Cao et al, 2007). After 6 h of ischemia followed by 24 h of reperfusion, we graded the neurologic impairment: score 0, no neurologic impairment; score 1, endoduction of right anterior limbs and no wholly stretch while left tail; score 2, circling toward right side when walking spontaneously; score 3, right-side lateriversion when walking; score 4, lack of spontaneous walk and some consciousness lost. We screened out the mice with a score of 0 or 4.
Measurement of Cerebral Water Content
We randomly took six mice for each group, put them to death after 24 h of reperfusion by decapitation, and levered the skull within 1 min to take out brain tissues. We then blotted up the water on the surface of the left brain with filter paper and took the humid weights (GW) on an electronic balance. Then we grilled them for 48 h at 110°C in an electrothermostatic blast oven and took their dry weights (DW). The cerebral water content was calculated by the formula cerebral water content % = (GWSYMBOL45}Symbol}11322GD)/GW · 100%.
Cerebral Infarction Size Assay
Mice were killed by excess narcosis with 2% pentobarbitol sodium. This was followed by immediate decapitation, and the brain tissue was removed and refrigerated at −70°C for 10 mins quick-freezing. Then, we obtained the procerebrum, and cut it into five corona brain slices of the same thickness (2 mm) from the frontal to the occipital pole. The slices were immediately placed in 2% tetrazolium chloride (Sigma Co., Shanghai, China)-PBS for protecting from light, incubated at 37°C for 30 mins, and fixed for 2 h with 4% paraformaldehyde. Normal brain tissue stained bright red, whereas infarction focus was pale white. We arranged the fixed brain slices in order, and measured the whole area of prosencephalon and cerebral infarction with Image Pro Plus 5.0 (IPP) image processing software. We also calculated the area according to the formula V = tx(A1 + A2+&An) (Majid et al, 2000), where V is infarction volume or prosencephalon volume, t is the thickness of slice, and A is the infarction size. We calculated the cerebral infarction volume and prosencephalon volume, as well as the volume ratio of cerebral infarction (cerebral infarction volume/prosencephalon volume).
Determination of Serum Levels of TNF-α, IL-6, and HSP60
Serum levels of TNF-α, IL-6, and HSP60 were determined by ABC-ELISA (a sandwich enzyme-linked immunosorbent assay) according to the instructions provided with the kits (Beijing Jingmei Co., Beijing, China). The serum level of HSP60 was graded as high, medium, and low. Then two cases with high HSP60 level, three cases with medium HSP60 level, and three cases with low HSP60 level were subjected to the following experiments.
Determination of NF-κB Activity by Electrophoretic Mobility Shift Assay
Transfected human CD14/TLR4 HEK293 cells from ATCC (USA), which do not express TLR4, MD2, and CD14 receptors, react to TLR4-related ligands such as LPS, or activate NF-κB, were stored in our laboratory. The cells were stabilized as described previously (Yang et al, 2002), and then cultured in RPMI (GIBCO-BRL, Biobank, Korea) containing 10% calf serum (Invitrogen, Beijing, China), 1 mg/mL G418 and 400 U/mL hygromycin B (Calbiochem-Novabiochem GmbH, Beijing, China) at 37°C in an atmosphere containing 5% CO2. On 90% cell confluence, HSP60 or anti-HSP60 (1:200, StressGen, Beijing, China) was added and the cells were cultured for another 12 h. To observe the specificity of HSP60 to NF-κB activation, 100 mg/L LPS was used in the positive control cells, and rat anti-human TLR9 (eBioscience) or anti-TLR9 + HSP60 or IgG (an isotype control antibody, eBioscience) or anti-TLR4 (eBioscience) was used to stimulate cells. The cells were washed three times with PBS, and digested with 2 mmol/L EDTA in PBS after a 12-h culture. Electrophoretic mobility shift assay (Wang and Siow, 2000) was performed as follows: nucleoprotein was extracted from HEK293 cells, 10 μg nucleoprotein of each sample was incubated with the reaction buffer at room temperature for 15 mins, then 32P-labeled oligonucleotide (5‘-GGGGACT TTCC-3’; Life Technologies, Gaithersburg, MD, USA), which binds to NF-κB, was added in the reaction buffer and incubated for 15 mins. After incubation at 25°C for 20 mins, the reaction mixture was subjected to 6% nondegeneration Polyacrylamide gel electrophoresis. Autoradiography was performed at −70°C. Finally the images were analyzed using BioRad Image Analyzer and the results were expressed as optical density.
Statistical Analysis
Measurement data are shown as mean ± s.d. or percentages. One-way ANOVA and Student-Newman-Keuls test in post hoc tests were used to analyze differences between the three groups. Pearson correlation analysis was used to reveal the correlation among TLR4 expression, NIHSS, and mRS, and the correlation among TLR4 expression, TNF-α, and IL-6. SPSS11.5 software was used in all statistical analyses. A P-value < 0.05 was deemed statistically significant.
Results
Baseline Data of Subjects
Demographic and clinical data of subjects are summarized in Table 1.
Correlation of TLR4 Expression, 10-day NIHSS, and 3-month mRS
Flow cytometry results showed low TLR4 expression on lymphocytes and neutrophils (< 3%), and there was no significant difference in TLR4 expression among the ACI, TIA, and control groups. Toll-like receptor 4 was mainly expressed on monocytes (ACI, 76.33% ± 18.65%; TIA, 46.24% ± 13.26%; control, 29.86% ± 12.54%) and the expression level was significantly higher in the ACI group than the control and TIA groups (P < 0.0001), and was also significantly higher in the TIA group than the control group (P < 0.0001), suggesting a marked increase in TLR4 expression on PBMs from ACI patients (Figure 1). Correlation analysis showed a positive correlation among TLR4 expression on PBMs, 10-day NIHSS score ((range: 9 to 31, median: 17); r = 0.799; P < 0.0001), and 3-month mRS ((range: 1 to 5, median: 4); r = 0.728; P < 0.0001), suggesting that TLR4 relates to the severity of ACI.
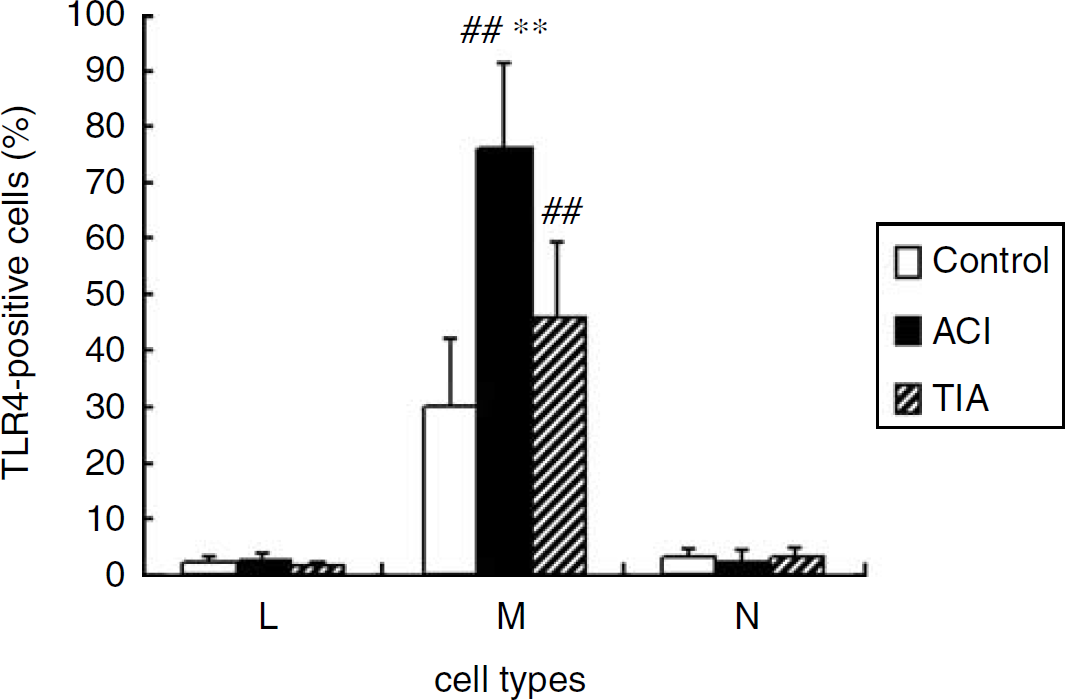
Comparison of TLR4 expression frequencies on PBMs among the control, ACI, and TIA groups. The PBMs were immunostained with TLR4 monoclone antibody. Ten thousand cells were analyzed by flow cytometry. L, lymphocytes; M, monocytes; N, neutrophilic granulocytes. Values represent mean ± s.d. **P < 0.0001 versus the control groups. **P < 0.0001 versus the TIA groups.
Expression of TLR4mRNA and MyD88mRNA
The results showed that the relative expression value of TLR4mRNA was 1.89 ± 0.36 in the ACI group, a figure significantly higher than that in the TIA group (0.85 ± 0.17) and the control group (0.36 ± 0.07) (P < 0.0001). The value was significantly higher in the TIA group than the control group (P < 0.0001). The results were coincident with those of TLR4 expression in PBMs (Figure 2). Correlation analysis showed that the relative expression value of TLR4mRNA significantly positively correlated with 10-day NIHSS (r = 0.816; P < 0.0001) and 3-month mRS (r = 0.771; P < 0.0001). Unlike TLR4mRNA expression, MyD88 mRNA expression differed insignificantly among the ACI (0.43 ± 0.05), TIA (0.47 ± 0.03), and control groups (0.46 ± 0.04; P > 0.05), suggesting that TLR4 may participate in cerebral ischemic injury through an MyD88-independent signal pathway.
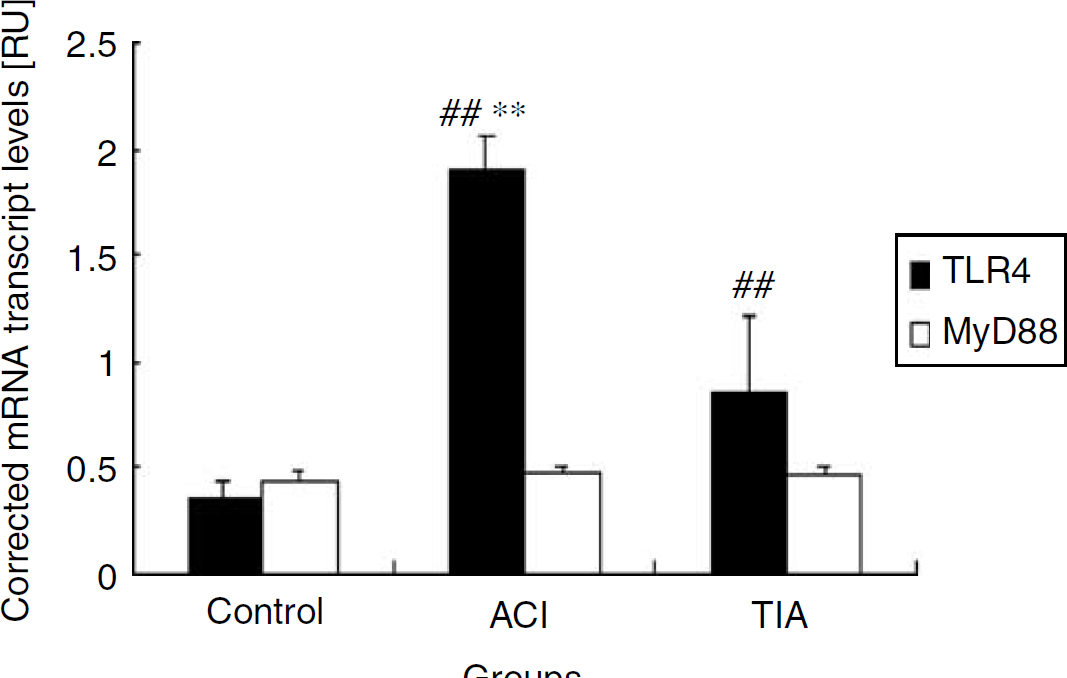
Expression of TLR4 mRNA and MyD88 mRNA on PBMs in the ACI, TIA, and control groups. Reverse transcriptase-PCR amplification results were normalized against GAPDH. Values represent mean ± s.d. **P < 0.0001 versus the control groups. **P < 0.0001 versus the TIA groups.
TLR4 May Participate in Cerebral Ischemic Injury Through an MyD88-Independent Signal Pathway
As MyD88 mRNA differed insignificantly between the cerebral infarction group and the control group, to elucidate the intermediate or downstream signal pathway by which TLR4 contributes to cerebral infarction, we observed cerebral ischemia/reperfusion injury in MyD88 gene knockout mice. As shown in Figure 3, compared with wild-type mice, 6 h of cerebral ischemia followed by 24 h of reperfusion did not significantly change cerebral edema, cerebral infarction area, and neurologic impairment scores in MyD88 gene knockout mice, suggesting that TLR4 may contribute to cerebral ischemia/reperfusion injury through an MyD88-independent signal pathway.
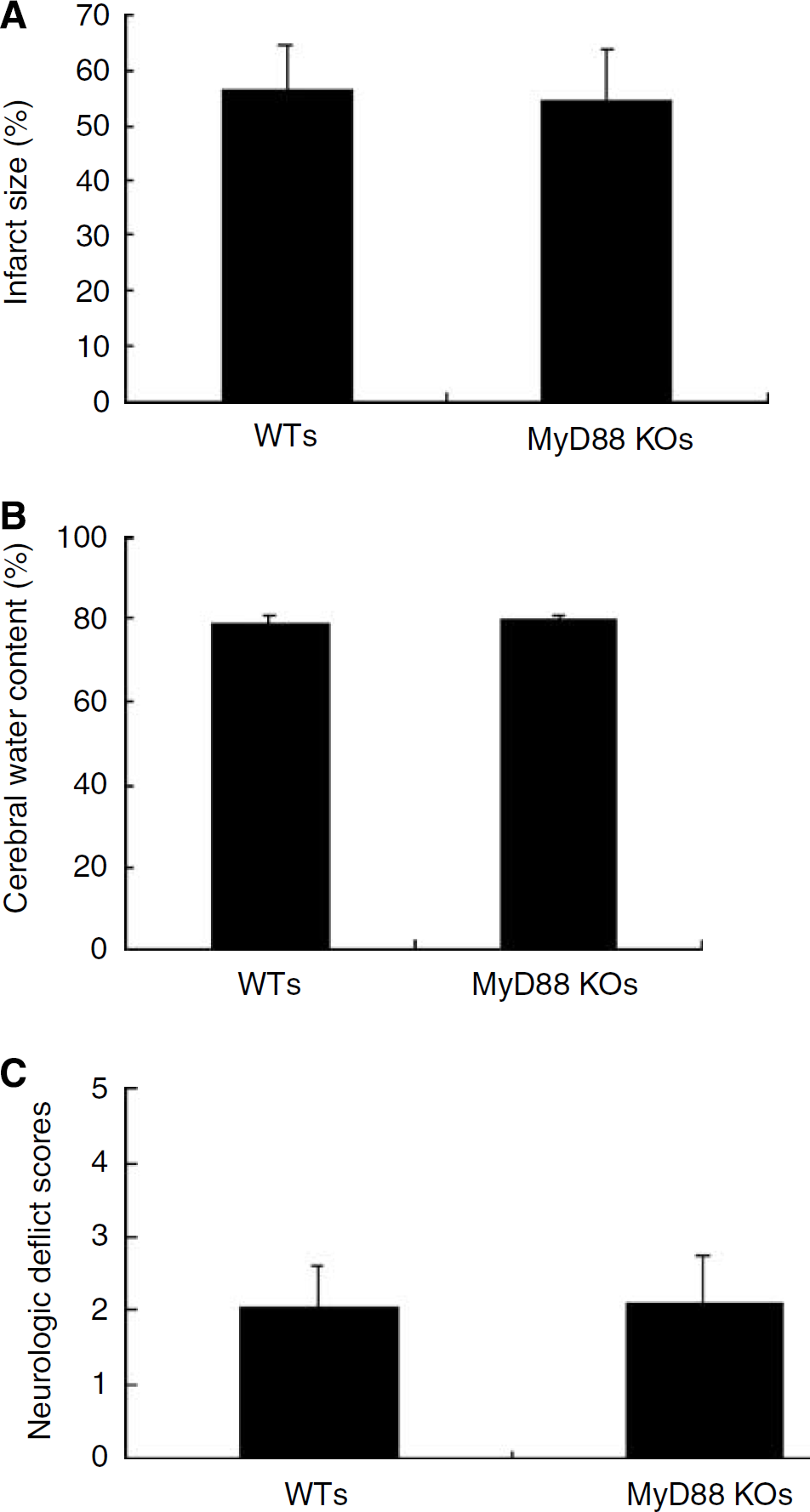
Toll-like receptor 4 may participate in cerebral ischemic injury through an MyD88-independent signal pathway. (
Serum Levels of TNF-α and IL-6 and their Correlation with TLR4 Expression
The results showed significant increases in serum levels of TNF-α and IL-6 in the ACI group, compared with the TIA and control groups (P < 0.0001). Also, the level was significantly higher in the TIA group than the control group (P < 0.0001) (Figure 4). Correlation analysis showed a positive correlation of TLR4 expression in PBMs and mRNA expression with serum levels of TNF-α and IL-6 in the ACI group, with correlation coefficients as follows: TLR4 expression in PBMs and TNF-α, 0.876; TLR4 expression in PBMs and IL-6, 0.783 (P < 0.001); TLR4mRNA and TNF-α, 0.732; TLR4mRNA and IL-6, 0.821 (P < 0.001).
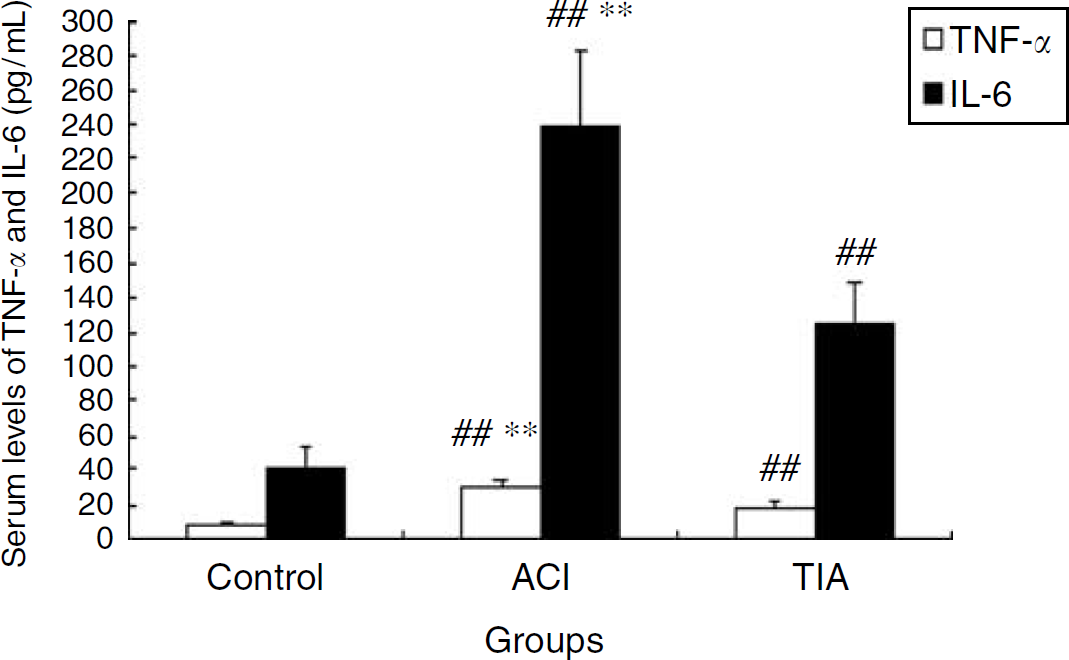
Serum levels of TNF-α and IL-6 (pg/mL) measured by ELISA in the ACI, TIA, and control groups. The TNF-α and IL-6 levels were higher in the ACI and TIA groups than the control group. Values represent mean ± s.d. **P < 0.0001 versus the control groups. **P < 0.0001 versus the TIA groups.
Serum HSP60
The HSP60 level was significantly higher in the ACI group than the TIA and control groups. Low levels of HSP60 were detected in only 10 patients of the control group. The HSP60 level was significantly higher in the TIA group than the control group (Figure 5).
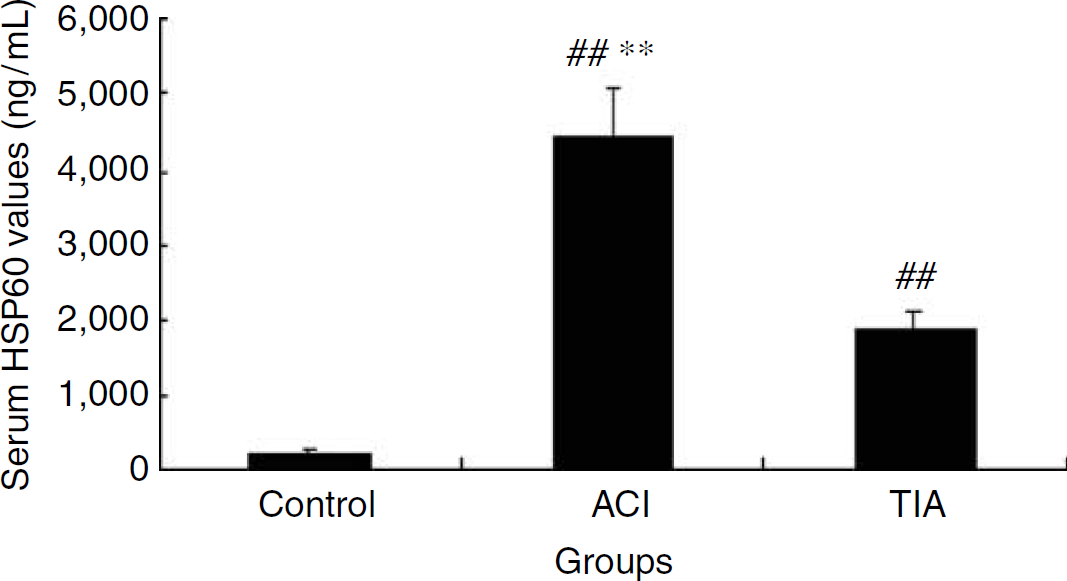
Serum HSP60 levels measured by ELISA. The HSP60 levels were higher in ACI and TIA groups than the control group. Values represent mean ± s.d. **P < 0.0001 versus the control groups. **P < 0.0001 versus the TIA groups.
Activation of NF-κB in CD14/TLR4-transfected HEK293 Cells by Serum HSP60
Serum HSP60 was significantly increased in the ACI and TIA groups. To investigate the possible ligands for TLR4, we detected whether the increased HSP60 can activate CD14+ /TLR4+ HEK293 cells and lead to NF-κB activation. Serum with the highest level of HSP60 was drawn from two patients, serum with moderate level of HSP60 was drawn from three patients, and serum with the lowest level of HSP60 was drawn from three patients of the TIA and ACI groups. Then, CD14+/TLR4+ HEK293 cells were stimulated with the sera. In western blotting, HSP60-specific monoclonal antibody was used to detect HSP60 specificity (data not shown). Lipopolysaccharide was used as the positive control. As shown in Figure 6, sera from both ACI and TIA groups significantly induced the activation of NF-κB in CD14+/TLR4+ HEK293 cells, but sera from the control group did not induce the activation of NF-κB. Moreover, such activation was significantly inhibited by anti-HSP60 antibody and anti-TLR4. Lipopolysaccharide significantly induced the activation of NF-κB, but anti-HSP60 antibody did not inhibit such activation. In addition, anti-TLR9 antibody or isotype control antibody did not significantly inhibit NF-κB activation induced by sera from both ACI group.
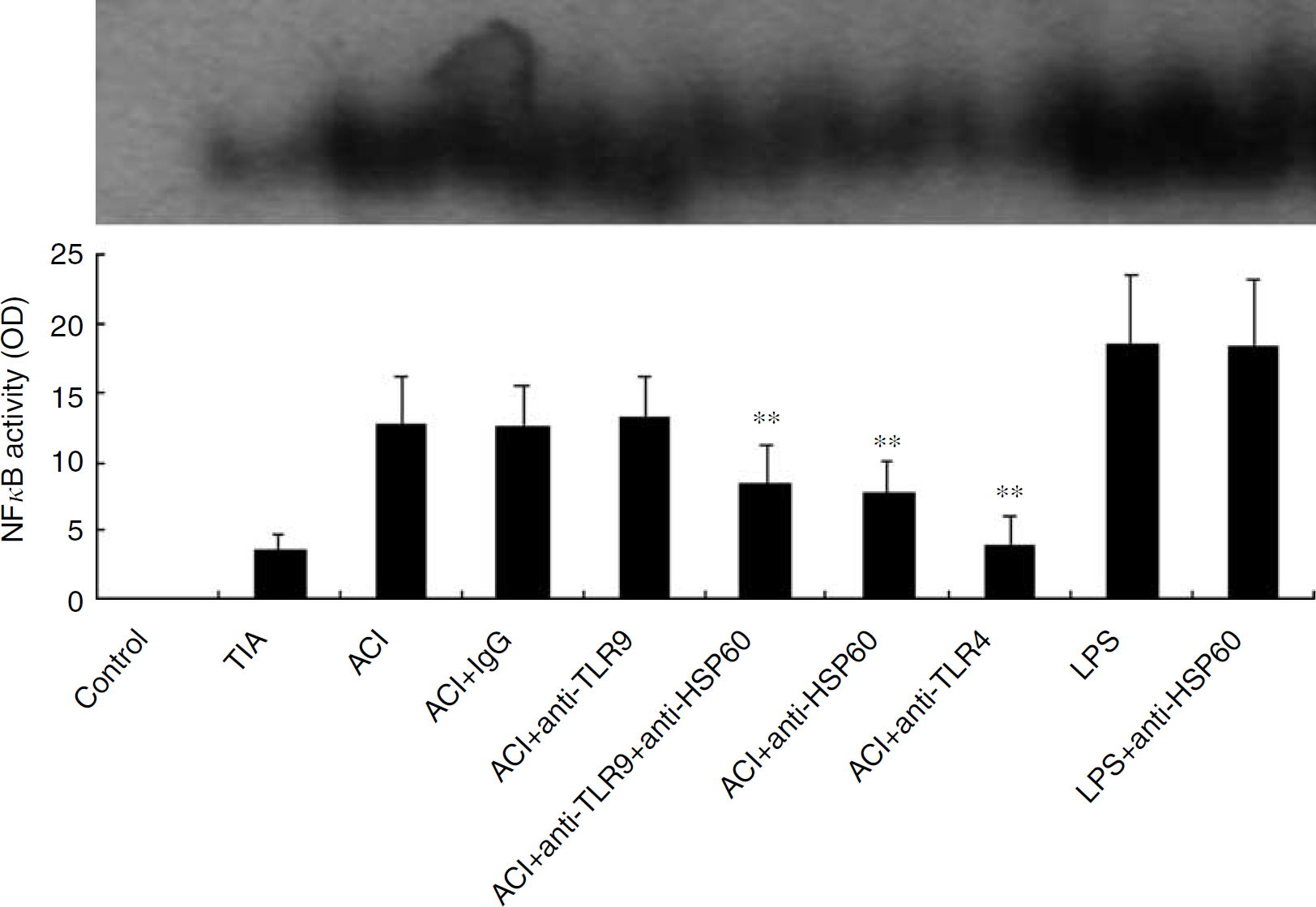
NF-κB activity assayed by electrophoretic mobility shift assay. The values represent mean ± s.d. of eight serum samples. **P < 0.001 versus the ACI group.
Discussion
To our knowledge, for the first time, we reported a significant increase of TLR4 expression on PBMs from ACI patients and a correlation of TLR4 expression with downstream inflammatory factors TNF-α and IL-6, as well as with the severity of stroke. We found a significant increase in peripheral HSP60 early after ACI, which may be one of the inflammatory injury mechanisms that induce endogenous ligands for TLR4 to participate in cerebral infarction. Interestingly, no significant change was observed in MyD88, a signal downstream of TLR4 in PBMs from ACI patients. It was also found that cerebral ischemia/reperfusion injury differed insignificantly between wild-type mice and MyD88 gene knockout mice, suggesting that TLR4 induces downstream signal molecules through an MyD88-independent signal pathway.
In the central nervous system, TLR4 is mainly expressed on oligodendrocytes, as well as on astrocytes, but not on neurons (Lehnardt et al, 2003a). Toll-like receptor 4 plays a key role in the innate immunity of the central nervous system (Lehnardt et al, 2002b). Lin et al (2005) showed that polymorphisms of TLR4 C119A gene significantly correlate with ischemic stroke, suggesting that TLR4, as a key genetic factor, participates in cerebral ischemic stroke. Tang et al (2007) recently showed TLR4 expression on neurons and the important role of TLR4 in inflammatory injury and functional deficits after cerebral ischemia. We previously showed (Cao et al, 2007) substantial reductions in the cerebral infarction area, nervous functional impairment scales, and cerebral inflammatory changes in TLR4 gene knockout mice, which suggest that TRL4 plays an important role in inflammatory injury after cerebral infarction. Both central and peripheral inflammatory injuries occur after cerebral infarction. Immediately after cerebral infarction, intense peripheral inflammatory reactions may occur, including activation of inflammatory cells and production of high levels of inflammatory cytokines. Because of injury to the blood—brain barrier after cerebral infarction, activated mononuclear macrophages enter the brain issue, causing inflammatory injury and intensified cerebral injury. Here we showed that TLR4 expression was significantly increased on PBMs, and inflammatory cells were activated at acute stages of cerebral infarction, and that these changes positively correlated with the levels of inflammatory cytokines, NIHSS, and mRS, showing that TLR4 participates in the inflammatory injury mechanism of cerebral infarction and may be one of the peripheral mechanisms of inflammatory injury after cerebral infarction. It is hypothesized that PBMs with upregulation of TLR4 expression may lead to secondary injury when they penetrate the blood—brain barrier, thus aggravating brain injury and neural functional deficits. Further investigations are needed to verify our hypothesis. In addition, TLR4 expression on PBMs positively correlated with the severity of stroke, suggesting TLR4 to be a predictor of ACI. In addition, to investigate whether reperfusion after thrombolysis influenced inflammatory reaction, we analyzed the inflammatory parameters (TLR4, TNF-α, IL-6) in the two patients who underwent thrombolysis and other patients who did not undergo thrombolysis, but found no significant differences in this regard (data not shown). Hence, we concluded that reperfusion after thrombolysis does not influence inflammatory reaction. However, this is not supported by literature. Moreover, the small sample size of this study does not support the conclusion either. Further work is required to elucidate whether thrombolysis ameliorates neurologic impairment through relieving inflammatory injury.
There are two classical TLR4 signal pathways, namely, MyD88-dependent and independent pathways. In the MyD88-dependent signal pathway, the downstream signal molecules IRAK and TRAF6 are activated, leading to NF-κB activation and production of inflammatory factors, such as TNF-α, IL-6, and IL-1 (Akira, 2006). Interestingly, in this study, Reverse transcriptase-polymerase chain reaction results did not show a significant change in MyD88mRNA on PBMs from ACI patients when compared with the control and TIA groups, suggesting the possible role of TLR4 in inflammatory injury of cerebral infarction through an MyD88-independent signal pathway. The findings are in agreement with other studies (Zhai et al, 2004). It was found that compared with wild mice, there was no obvious change in brain edema, cerebral infarction area, or neurologic impairment scale after cerebral ischemia/reperfusion in MyD88 gene knockout mice. Therefore, we maintain that TLR4 does not participate in cerebral ischemia/reperfusion injury through the classical MyD88-dependent pathway.
Heat-shock protein 60 was shown to be an endogenous ligand for TLR4 (Ohashi et al, 2000). Accumulating evidence has showed that TLR4 and HSP60 play key roles in the progress of atherogenesis and atherosclerosis (Vink et al, 2002; Wick et al, 2004). Methe et al (2005) reported that HSP60 plays a key role in acute coronal artery syndrome as an endogenous ligand. We found here that sera from patients with increased HSP60 specifically activated CD14+/TLR4+HEK293 cells and led to NF-κB activation, and that anti-HSP60 antibody significantly inhibited such NF-κB activation, suggesting that HSP60 also participates in inflammatory injury after ACI as an endogenous ligand for TLR4. Some authors (Tsan and Gao, 2004) suggested that HSPs activate TLR4 with the help of LPS, and that HSP60 activates TLR4 because of LPS contamination or the action of other LPS-related ligands. However, we did not detect LPS in the sera from the control, TIA, and ACI groups (data not shown), and found that anti-HSP60 antibody insignificantly inhibited the activation of CD14+/TLR4+HEK293 cell by LPS. Hence, it is concluded that HSP60 acts as a ligand for TLR4, without the involvement of LPS. The present study preliminarily indicates that circulating HSP60 can directly activate TLR4 in ACI, a finding consistent with the report on coronal artery syndrome by Methe H. Nevertheless, it should be further investigated whether HSP60 is a ligand for TLR4 in inflammatory injury after ACI.
In summary, the present study preliminarily indicates significant upregulation of TLR4 expression on PBMs from ACI patients, which positively correlates with the expression of downstream inflammatory factors and the severity of stroke, suggesting upregulation of TLR4 expression to be one of the peripheral mechanisms of inflammatory injury after cerebral infarction. Peripheral blood monocytes with upregulation of TLR4 expression may lead to secondary injury when they penetrate the blood—brain barrier, thus aggravating brain injury and neural functional deficits. In addition, it is suggested that TLR4 may be a predictor for ACI. Coincident with other researches, we show that circulating HSP60 may participate in the peripheral mechanism of inflammatory injury after ACI as a ligand for TLR4, but not through the classical MyD88-dependent signal pathway.
Footnotes
The authors state no conflict of interest.