
Abstract
Stroke induced by middle cerebral artery occlusion leads to transiently increased progenitor proliferation in the subventricular zone (SVZ) and long-lasting striatal neurogenesis in adult rodents. Tumor necrosis factor-α (TNF-α) is upregulated in stroke-damaged brain. Whether TNF-α and its receptors influence SVZ progenitor proliferation after stroke is unclear. Here we show that the increased proliferation 1 week after stroke occurred concomitantly with elevated microglia numbers and TNF-α and TNF receptor-1 (TNF-R1) gene expression in the SVZ of wild-type mice. TNF receptor-1 was expressed on sorted SVZ progenitor cells from nestin-green fluorescent protein reporter mice. In animals lacking TNF-R1, stroke-induced SVZ cell proliferation and neuroblast formation were enhanced. In contrast, deletion of TNF-R1 did not alter basal or status epilepticus-stimulated cell proliferation in SVZ. Addition of TNF-α reduced the size and numbers of SVZ neurospheres through a TNF-R1-dependent mechanism without affecting cell survival. Our results provide the first evidence that TNF-R1 is a negative regulator of stroke-induced SVZ progenitor proliferation. Blockade of TNF-R1 signaling might be a novel strategy to promote the proliferative response in SVZ after stroke.
Introduction
Ischemic stroke, induced by middle cerebral artery occlusion (MCAO) in adult rodents, leads to increased proliferation of progenitor cells in the subventricular zone (SVZ) and migration of newly formed neuroblasts into the damaged striatum (for references, see Lindvall and Kokaia, 2008). Many of the stroke-generated neuroblasts differentiate into mature neurons with the phenotype of striatal projection neurons (Arvidsson et al, 2002; Parent et al, 2002). Striatal neurogenesis after stroke is long-lasting and continues during several months after the insult (Thored et al, 2006). Recent findings suggest increased progenitor proliferation and formation of immature neurons also in the human brain (Jin et al, 2006). From a clinical-therapeutic perspective, the neurogenic response from SVZ progenitor cells after stroke is probably insufficient and only a fraction of the dead neurons are replaced (Arvidsson et al, 2002). Optimization of neurogenesis after stroke will require identification of the molecular mechanisms regulating its various steps, that is, proliferation, survival, migration, differentiation, and functional integration. Overall cell proliferation in the SVZ is transiently increased 1 to 2 weeks after MCAO (Arvidsson et al, 2002; Jin et al, 2001). When supplied to the SVZ, many factors increase cell proliferation after stroke (Lindvall and Kokaia, 2008). However, the intrinsic regulation of stroke-induced proliferation is poorly understood, and no endogenous mechanism that suppresses the proliferative response in the SVZ has yet been identified.
Tumor necrosis factor-α (TNF-α) is a major player in many neurodegenerative diseases including stroke (Hallenbeck, 2002), its main source being activated microglia (Gebicke-Haerter, 2001). Tumor necrosis factor-α influences cell survival through the action on two different receptor subtypes, TNF-R1 and TNF-R2 (Wajant et al, 2003): TNF-R1 contributes to neuronal death, whereas TNF-R2 can be neuroprotective (Marchetti et al, 2004). We recently identified TNF-R1 signaling as a negative regulator of progenitor proliferation in hippocampal neurogenesis (Iosif et al, 2006). Cell proliferation in the dentate subgranular zone (SGZ) was enhanced in TNF-R1−/− mice both under basal conditions and after status epilepticus (SE).
Whether TNF-R1 signaling influences progenitor proliferation in SVZ after stroke is unclear. Neurosphere cultures from rat SVZ express TNF-R1 (Widera et al, 2006) and indirect evidence suggests that TNF-α might stimulate SVZ cell proliferation. Intraventricular delivery of TNF-α in rats increased bromodeoxyuridine (BrdU) incorporation in SVZ cells (Wu et al, 2000). Widera et al (2006) reported that TNF-α increased volume, BrdU incorporation, and total cell numbers in neurosphere cultures from intact rat SVZ. Finally, Katakowski et al (2007) showed upregulation of TNF-α-converting enzyme in SVZ after stroke and decreased stroke-induced SVZ proliferation after infusion of an inhibitor of this protease.
Here we have used mouse models with loss of TNF-R1 and TNF-α function, combined with in vitro and in vivo analyses. The main objectives were first to determine whether signaling through TNF-R1 influences SVZ progenitor proliferation after stroke induced by 40 mins MCAO. This insult, which causes damage to striatum and cerebral cortex and massive microglia activation, is associated with TNF-R1 and TNF-α upregulation in the ischemic hemisphere (Lambertsen et al, 2005, 2007). The second objective was to explore if the enhanced SVZ progenitor proliferation caused by SE, that is, a pathologic condition that does not lead to extensive striatal and cortical damage, is affected by TNF-R1 signaling. The third objective was to analyze whether TNF-R1 and TNF-α are expressed in mouse SVZ progenitor cells and if gene levels are altered after stroke and, finally, to establish any TNF-R1-mediated effect on SVZ cell proliferation in vitro.
Materials and methods
Animals and Surgery
All experimental procedures followed guidelines set by the Malmö-Lund Ethical Committee. We generated TNF-R1−/− mice by crossing TNF-R1/R2−/− to wild-type C57BL/6 mice (B&K Universal, Stockholm, Sweden). The TNF-R1/R2−/− mice (Peschon et al, 1998), back-crossed for five generations into wild-type C57BL/6 mice, were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Interbreeding of heterozygous offspring was followed by selection of appropriate genotypes using PCR. TNF-α−/− mice (Pasparakis et al, 1996), back-crossed for 10 generations into wild-type C57BL/6 mice, were purchased from Jackson Laboratories. Wild-type C57BL/6 mice were used as controls.
A total of 33 TNF-R1−/−, 12 TNF-α−/−, and 45 wild-type adult mice, weighing 22 to 24 g, were used for in vivo and in vitro studies. In addition, five adult nestin-green fluorescent protein (GFP) mice (G Enikolopov, Cold Spring Harbor Laboratory, NY, USA), weighing 28 to 30 g, were used for progenitor sorting. Animals were housed separately under 12 h light/dark conditions with ad libitum access to food and water.
Nineteen mice (TNF-R1−/−, n = 10; wild type, n = 9) were anaesthetized with halothane and implanted with a twisted, insulated stainless-steel stimulating/recording electrode (Plastics One, Roanoke, VA, USA) unilaterally into the ventral hippocampal CA1-CA3 region (coordinates: 2.9 mm caudal and 3.0 mm lateral to bregma, 3.0 mm ventral from dura, tooth bar at −3.3 mm).
Induction of Stroke
The intraluminal filament model was performed on 23 mice (TNF-R1−/−, n = 8; wild-type, n = 4; TNF-α−/−, n = 5; wild-type, n = 6) (Hara et al, 1996). Briefly, right carotid arteries were exposed, external carotid was ligated, and temporary sutures were placed around the common and internal carotid arteries. A small incision was made in the external carotid artery and an 8-0 monofilament (Alcon Surgical Inc., Stockholm, Sweden) coated with silicone (Xantopren L, Heraeus, Germany) was advanced through the internal carotid artery until it blocked the blood flow in the middle cerebral artery. The filament was secured, wounds temporarily closed, and animals allowed to wake up. For reperfusion, mice were reanesthetized after 40 mins of occlusion and the filament was removed. Body temperature was maintained between 36.5°C and 37.5°C with a heating lamp during surgery and ischemia, and for 2 h thereafter. To assess whether the MCAO had been successful in individual mice, we performed a contralateral rotation test (Bederson et al, 1986) and a forelimb and hindlimb placing test (De Ryck et al, 1989) 2 h after the onset of reperfusion. Animals without motor impairment were excluded from the study. At 1 week after stroke, body weight did not differ between wild-type (28 ± 0.4 g), TNF-α−/− (28 ± 0.6 g), and TNF-R1−/− mice (27 ± 0.6 g), and the mortality rate was less than 10% in all groups without significant differences.
Induction of SE
Ten days after electrode implantation, 11 mice (TNF-R1−/−, n = 6; wild type, n = 5) were subjected to electrically induced SE (Iosif et al, 2006). Mice received 1 h of suprathreshold stimulation consisting of 10 secs trains of 1 ms biphasic square wave pulses at a frequency of 50 Hz. Stimulation was interrupted for 1 min every 10 mins to allow for EEG recording and measurement of afterdischarges (MacLab; AD Systems, Hastings, UK). After ending the stimulation, all mice exhibited self-sustained, continuous ictal hippocampal EEG activity and associated motor behavioral convulsions, categorized into partial and generalized seizures (Iosif et al, 2006). Behavioral convulsions and ictal EEG activity were arrested with pentobarbital (40 mg/kg intraperitoneal) at 2 h after stimulation offset.
Bromodeoxyuridine Administration
To label mitotic cells (Dolbeare, 1995), mice were administered the thymidine analogue BrdU (50 mg/kg, intraperitoneal), dissolved in potassium phosphate-buffered saline (KPBS), four times with a 2 h interval and were perfused 2 h thereafter. Animals were injected with BrdU at 7 days after MCAO or SE.
Immunohistochemistry
All animals received an overdose of sodium pentobarbital and were transcardially perfused with 100 mL of saline followed by 100 mL of ice-cold 4% paraformaldehyde in 0.1 mol/L KPBS. Brains were removed, postfixed overnight, and placed in 20% sucrose in 0.1 mol/L phosphate buffer for 24 h. Coronal sections (30 μm) were cut on a freezing microtome and stored in cryoprotective solution.
For double-label immunofluorescence with BrdU and the neuroblast marker doublecortin (Dcx) (Brown et al, 2003) or neuron-specific nuclear protein (NeuN), free-floating sections were denatured in 1 mol/L HCl for 30 mins at + 65°C, rinsed in KPBS, preincubated with 2% donkey, 2% horse, or 2% goat serum in 0.25% Triton X-100 in KPBS (T-KPBS) for 1 h, and incubated overnight with rat anti-BrdU antibody (1:100; Sigma, Stockholm, Sweden), goat anti-Dcx antibody (1:400; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), or mouse anti-NeuN primary antibodies (1:100; Chemicon, Temecula, CA, USA) at +4°C. Sections were then rinsed and incubated in darkness for 2 h with Cy3-conjugated donkey anti-rat (1:200; Jackson ImmunoResearch), biotinylated horse anti-goat (1:200; Vector Laboratories, Peterbourough, UK), or biotinylated horse anti-mouse secondary antibodies (1:200; Vector Laboratories), then incubated in Streptavidin Alexa Fluor 488 (1:200; Invitrogen, Stockholm, Sweden), mounted on glass slides, and coverslipped with glycerol-based mounting medium.
For staining of proliferating cell nuclear antigen (PCNA), which is a protein involved in DNA replication (Paunesku et al, 2001), phosphorylated histone H3 (p-H3), which labels cells in mitotic phase (Hendzel et al, 1997), and macrophage/microglia-specific protein Iba1, which identifies both activated and nonactivated forms of microglia (Imai and Kohsaka, 2002), free-floating sections were quenched with 3% H2O2 and 10% methanol in KPBS for 30 mins at room temperature, rinsed in KPBS, preincubated for 1 h with 2% horse serum in T-KPBS for PCNA and 2% goat serum in T-KPBS for p-H3 or Ibal. Sections were then incubated overnight at +4°C with mouse anti-PCNA antibody (1:1,000; Santa Cruz Biotechnology Inc.), rabbit anti-p-H3 antibody (1:400; Upstate, Charlottesville, VA, USA), and rabbit anti-Iba1 antibody (1:1,000; Wako Chemicals GmbH, Neuss, Germany), rinsed and incubated with biotinylated horse anti-mouse IgG antibody (1:200; Vector Laboratories) for PCNA or biotinylated goat anti-rabbit IgG antibody (1:200; Vector Laboratories) for p-H3 and Iba1. Sections were incubated with the avidin—biotin—peroxidase complex (Elite ABC kit, Vector Laboratories) for 1.5 h, treated with diaminobenzidine (0.5 mg/mL) and 3% hydrogen peroxide, and finally rinsed in KPBS, mounted on glass slides, and dehydrated in ethanol, before being coverslipped with Pertex mounting medium.
For Fluoro-Jade staining, free-floating brain sections were mounted on glass slides and allowed to dry overnight. They were then rehydrated, preheated with 0.06% potassium permanganate for 15 mins, rinsed in distilled water, incubated in 0.01% Fluoro-Jade working solution (Histo-Chem, Jefferson, AR, USA) for 30 mins, rinsed in distilled water, immersed in xylene, and cover-slipped (Schmued et al, 1997).
For terminal deoxynucleotidyl transferase-mediated fluorescein-dUTP nick-end labeling (TUNEL), free-floating sections were mounted on glass slides and allowed to dry. Sections were then preheated with 4% paraformaldehyde for 20 mins, methanol for 30 mins, proteinase K (10 μg/mL in KPBS) for 6 mins, 4% paraformaldehyde for 5 mins, and ice-cold 0.1% Triton X-100 in 0.1% sodium citrate for 2 mins, with KPBS rinses between each step. Subsequently, sections were incubated in the dark at + 37°C for 60 mins in terminal deoxynucleotidyl transferase buffer, containing 17 μL terminal deoxynucleotidyl transferase enzyme solution and 150 μL TUNEL label solution with fluorescein-conjugated dUTP (Boehringer Mannheim, Mannheim, Germany). Sections were counterstained with Hoechst 33342 (Molecular Probes, Stockholm, Sweden), 10 μg/mL, for 10 mins in darkness (Whiteside et al, 1998). Slides were coverslipped with glycerol-based mounting medium.
Microscopical Analysis
All assessments were performed by an observer blind to treatment conditions. Immunostainings were examined using an Olympus AX-70 fluorescence light microscope. Stereological estimations of the total number of BrdU + and PCNA+ cells in the SVZ were performed using the optical fractionator method (Gundersen and Jensen, 1987; West et al, 1991). Briefly, four coronal sections throughout the SVZ, located 0.74, 0.50, 0.14, and 0.02 mm anterior to bregma, were analyzed ipsilaterally and contralaterally using an Olympus BH-2 microscope with a × 100 objective, CCD-IRIS color video camera, and CAST-GRID software (Olympus, Ballerup, Denmark). For systematic sampling, the frame area was set at 1,491.7 μm2 with a step length of 40 μm in X and Y directions, and the optical dissector constituting a 5 μm thick fraction of the total section thickness. Infarct volume was measured using stereological equipment in four NeuN- and 4 Fluoro-Jade-stained sections throughout the striatum (+0.74, +0.50, +0.14, and +0.02 mm from bregma) as previously described by Thored et al (2006). The size of the SVZ was measured at the same rostrocaudal levels in BrdU/NeuN-stained sections. The density of Dcx+ cells in the rostral migratory stream was estimated by measuring the area of the rostral migratory stream in a 30 μm thick coronal section using stereological equipment. Numbers of Dcx+ cells were counted using an Olympus BH-2 microscope with a × 40 objective in the same section. Numbers of migrating Dcx + cells in the stroke-damaged striatum were estimated in four BrdU/Dcx-stained sections in a 150 μm wide column adjacent to SVZ. The column was subdivided into three equally sized, 50 μm wide columns. Numbers of Dcx+ cells in SVZ were estimated semiquantitatively: score 1, < 20%; 2, 20% to 50%; 3, > 50% of the SVZ area ipsilateral to damage is covered by Dcx+ cells. Owing to their low numbers, counts of BrdU + /Dcx +, p-H3 +, Iba1 +, and TUNEL + cells in the SVZ are given as cells per section. Colocalization of BrdU and Dcx was validated in a confocal laser scanning microscope (Leica). TUNEL + cells were counted if a clear apoptotic nuclear morphology (shrunken or fragmented nuclei), as assessed by Hoechst staining, was identified. Cell counts represented in Figures 1,2,4, and 5 are from SVZ on the side ipsilateral to stroke or electrically induced SE.
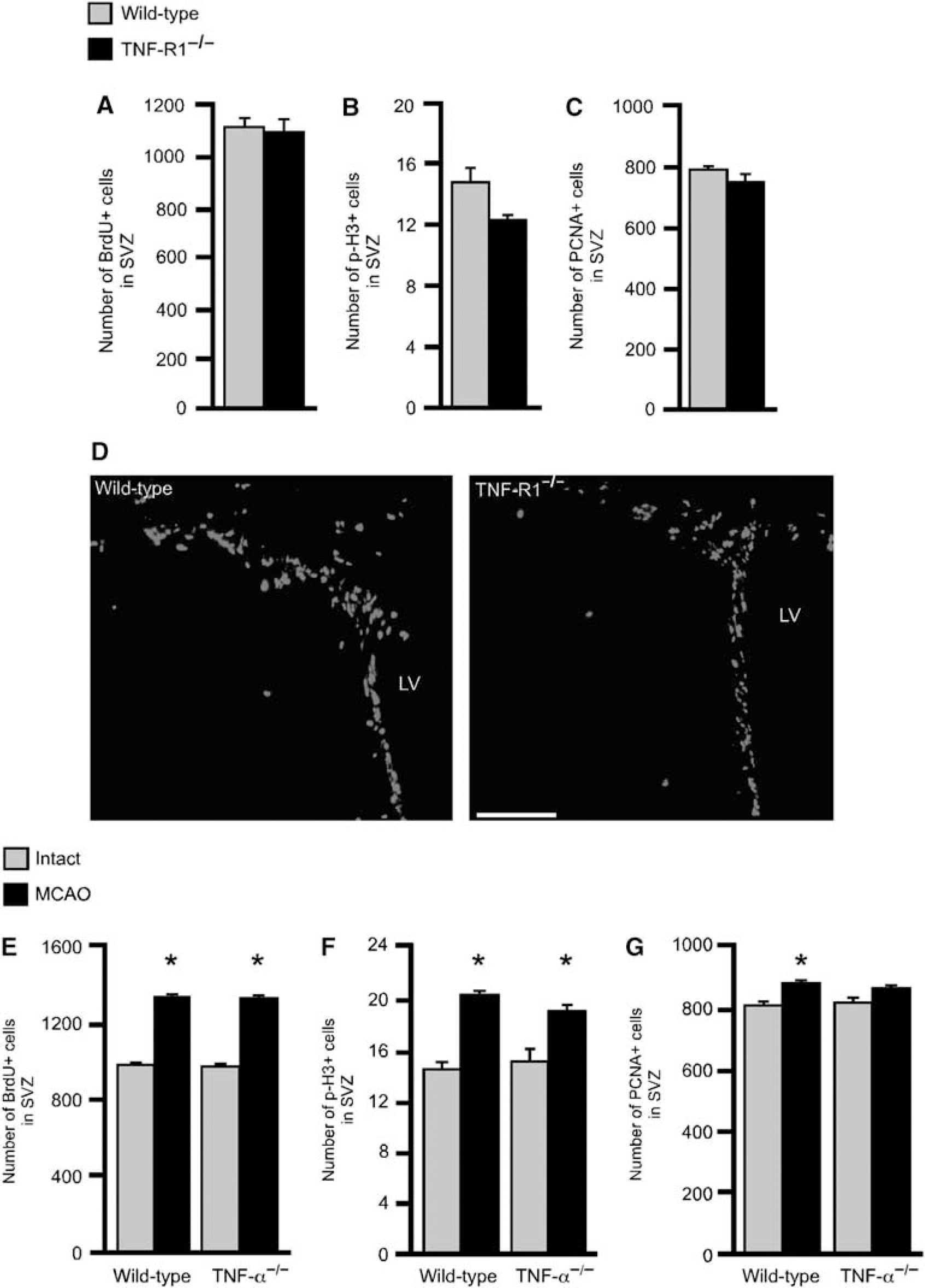
Deletion of TNF-R1 does not affect basal progenitor proliferation in SVZ and deletion of TNF-α does not alter progenitor proliferation in SVZ under basal conditions or after stroke. (
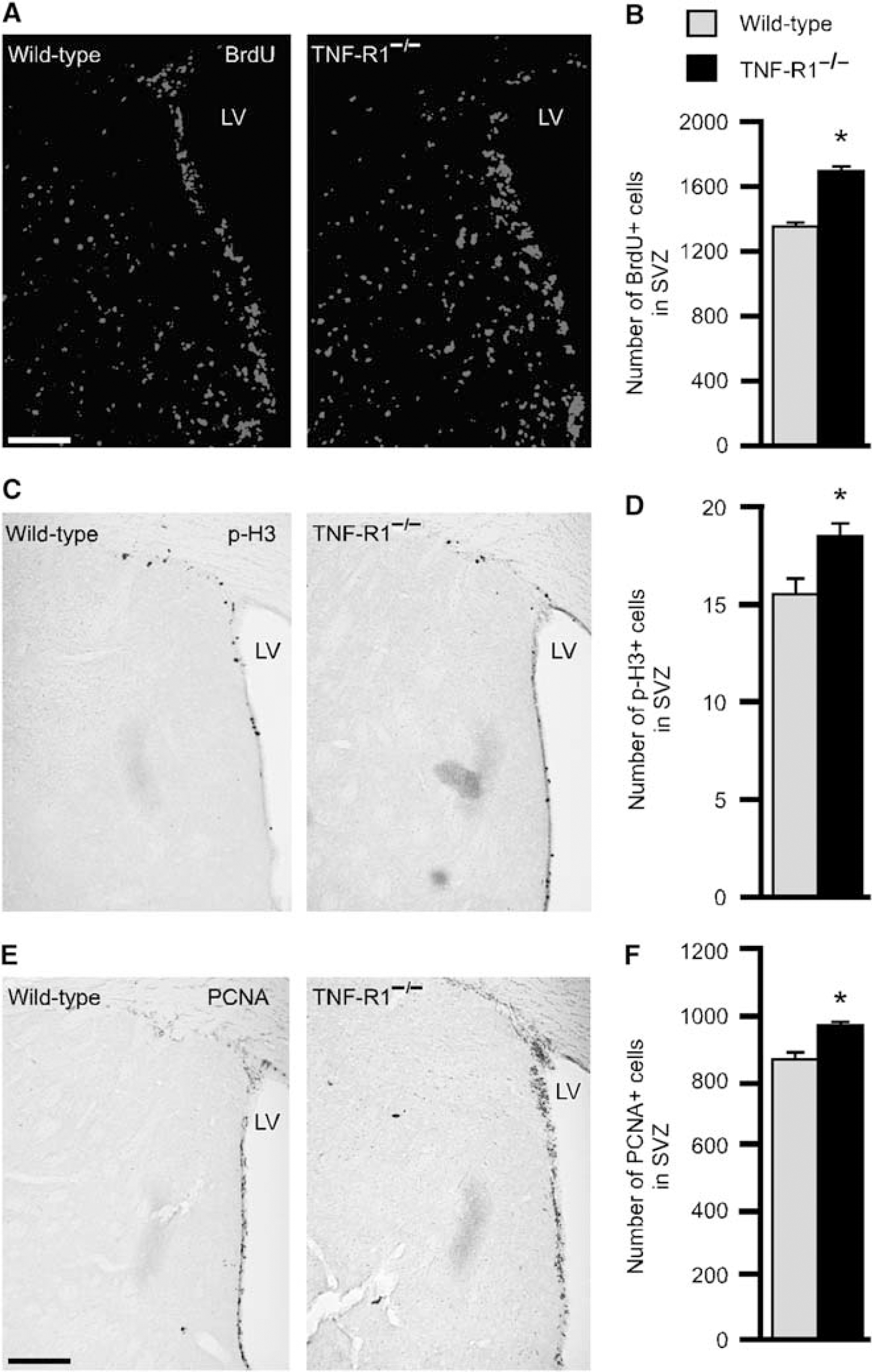
Deletion of TNF-R1 enhances the increased progenitor proliferation in SVZ after stroke. Photomicrographs showing the distribution of BrdU + (
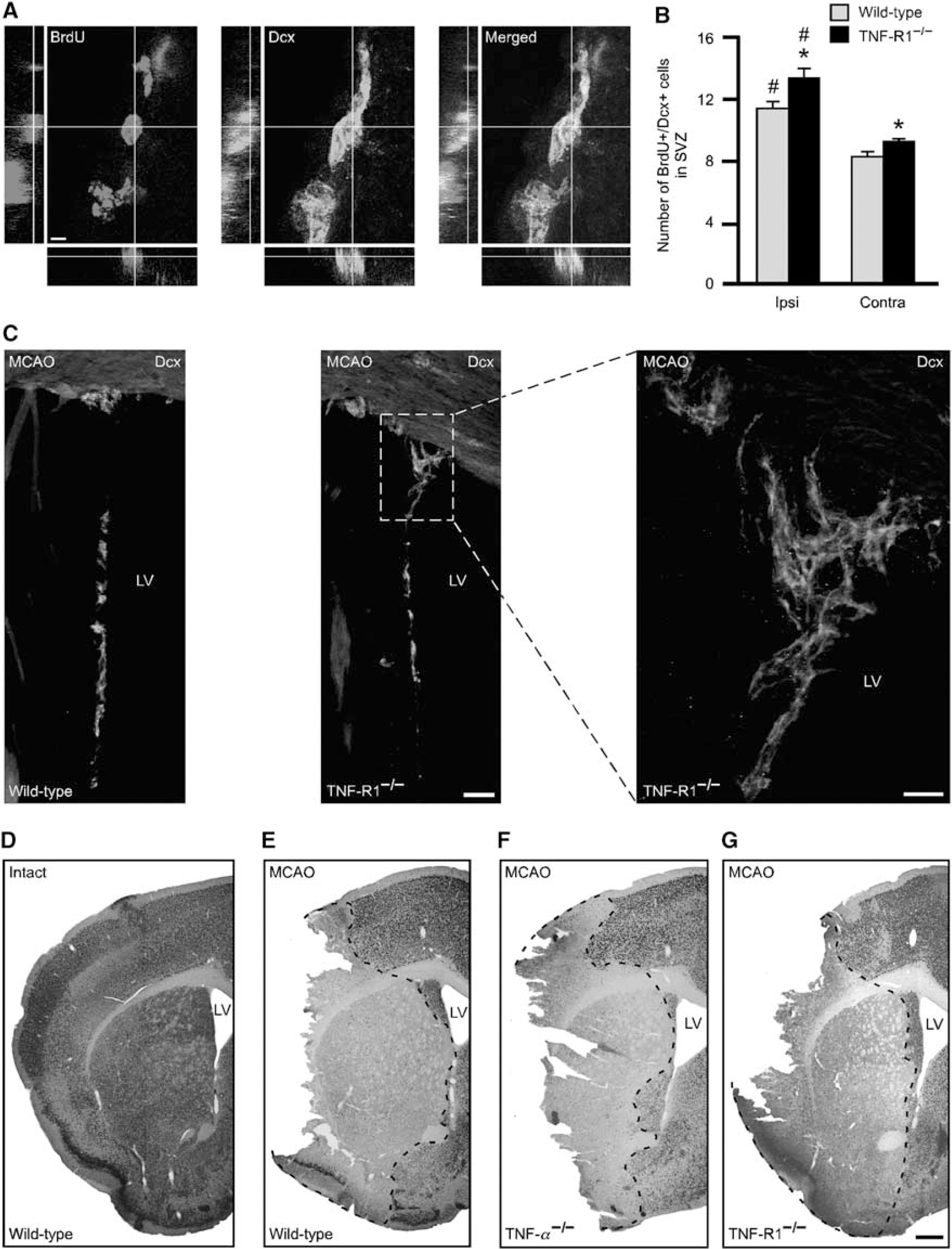
Deletion of TNF-R1 enhances the formation of neuroblasts in the SVZ after stroke but the size of ischemic injury is unchanged. (
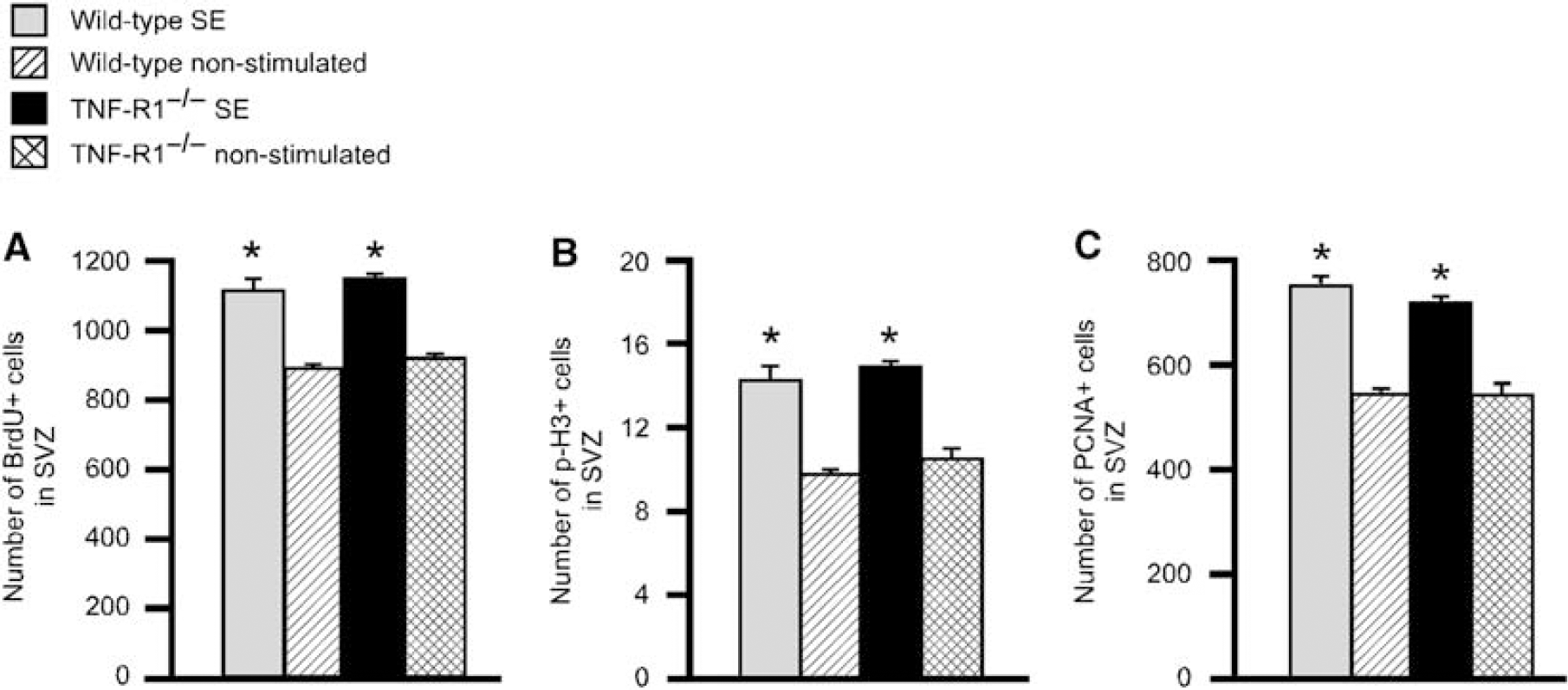
Deletion of TNF-R1 does not affect the increased progenitor proliferation in SVZ after SE. (
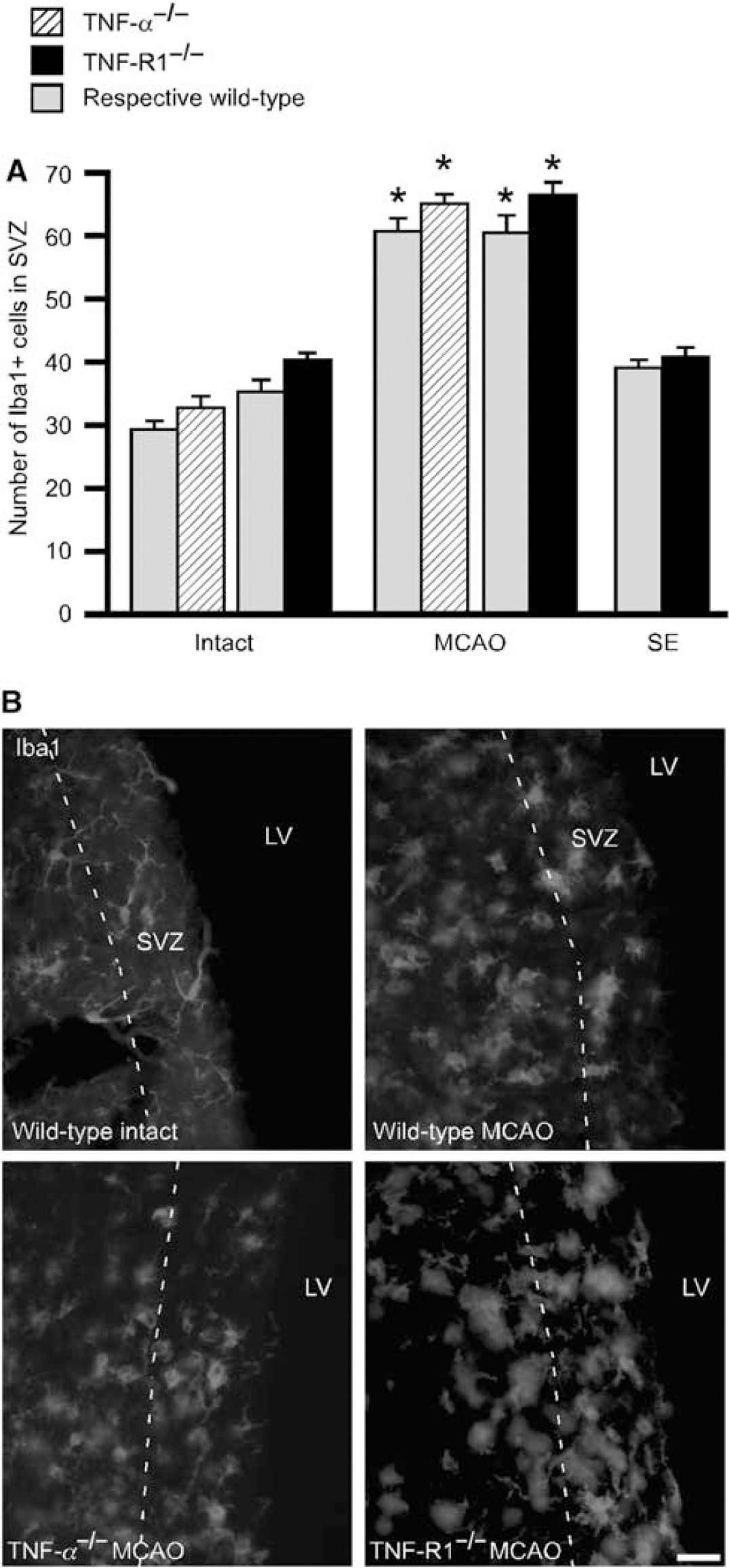
Stroke but not SE increases microglia numbers in SVZ. (
Dissection and Cell Culture
Animals were deeply anesthetized with halothane and killed with cervical dislocation. Brains were removed, placed in ice-cold L-15 (Gibco, Stockholm, Sweden), and cut into 1 mm thick coronal slices. For gene expression studies, slices were immediately transferred to RNAlater (Ambion, Stockholm, Sweden). The SVZ was subdissected from 1 mm sections using microscissors and processed for cell culture, cell sorting, or gene expression studies. The SVZ was enzymatically dissociated in HBSS with 0.015 mol/L HEPES, 5.4 mg/mL
Neurospheres were grown in DMEM/F12 supplemented with B27, 0.6% glucose (Sigma), 2 mmol/L
Neurospheres were passaged and cells grown adherent in proliferative medium without heparin on poly-
Cell Sorting, Reverse Transcription-PCR (RT-PCR), and Quantitative PCR
GFP + and GFP- cells from nestin-GFP animals were sorted on fluorescence-activated cell sorter (FACS DiVa; Becton Dickinson) directly into RLT lysis buffer (Qiagen, Solna, Sweden). An initial gate based on forward and side scatter was set to exclude debris and cell aggregates; 7AAD was used to exclude dead cells. A sorting gate was set around the main GFP + population, at least 1 log higher than GFP- controls.
RNA from nestin-GFP + and GFP- sorted cells, intact or stroke-damaged SVZ and striatum as well as from primary and expanded neurospheres was isolated using RNAeasy micro (Qiagen) with DNase treatment. RNA was reverse transcribed using oligoDT primers and superscript-II (Invitrogen). Semiquantitative PCR was performed as described elsewhere (Iosif et al, 2006).
Quantitative RT-PCR was performed with TaqMan universal master mix and TaqMan Gene expression assays were performed for GAPDH (Mm99999915_g1), TNF-α (Mm00443258_m1), and TNF-R1 (Mm00441889_m1). 100 ng of cDNA was used for each quantitative PCR and all experiments were run in triplicate. cDNA input was normalized to GAPDH. Relative gene expression was calculated using the ΔΔCT method.
Statistical Analysis
Comparisons between animal groups were performed using unpaired Student's t-test. Side differences, ipsilateral vs contralateral to the ischemic damage were assessed with paired Student's t-test. Comparisons between neurosphere numbers and size were performed using one-way analysis of variance with Bonferroni—Dunn post hoc test. Comparisons of semi-quantitative scores were analyzed with nonparametric Mann-Whitney U-test. Data are presented as means ± s.e.m., and differences are considered significant at P < 0.05.
Results
Deletion of TNF-R1 and TNF-α Does Not Affect Basal Progenitor Proliferation in SVZ
We first studied whether absence of TNF-R1 influenced SVZ cell proliferation in the intact brain. Three different proliferation markers were analyzed using immunohistochemistry: BrdU, p-H3, and PCNA. No differences in the numbers of BrdU +, p-H3 +, or PCNA+ cells in SVZ were detected between TNF-R1−/− and wild-type mice (Figures 1A–1C).
We also determined whether deletion of TNF-α caused alterations of cell proliferation in SVZ. We did not find any significant differences between TNF-α−/− and wild-type mice in the number of BrdU +, p-H3 +, or PCNA + cells under basal conditions (Figures 1E–1G).
Deletion of TNF-R1 but not TNF-α Enhances the Increased Progenitor Proliferation in the SVZ After Stroke
We wanted to explore whether after stroke, causing damage to the adjacent striatum and microglia activation, signaling through TNF-R1 regulates cell proliferation in SVZ. Wild-type and TNF-R1−/− mice were subjected to 40 mins MCAO and cell proliferation was analyzed 1 week thereafter. Stroke gave rise to higher numbers of BrdU+ cells in the SVZ bilaterally in both animal groups (Figure 2B) compared with intact animals (Figure 1A). Interestingly, after stroke, mice lacking TNF-R1 had elevated numbers of BrdU +, p-H3 +, and PCNA + cells in the SVZ compared with wild-type mice (Figure 2). There was no difference between wild-type and TNF-R1−/− mice in SVZ size at 1 week after MCAO (data not shown).
We then determined whether the enhancement of stroke-induced cell proliferation in the SVZ, caused by deletion of TNF-R1, also led to increased neuroblast generation. As a neuroblast marker we used Dcx. Bromodeoxyuridine had been injected 8, 6, 4, and 2 h before the animals were perfused at 1 week after MCAO. The number of BrdU + /Dcx + neuroblasts was significantly higher in the SVZ bilaterally in TNF-R1−/− as compared with wild-type mice (Figure 3B). In both TNF-R1−/− and wild-type mice, the number of BrdU + /Dcx + cells was higher in the SVZ on the stroke-damaged side. We obtained no evidence that the higher number of proliferating cells in the SVZ of TNF-R1−/− mice was because of decreased apoptotic cell death. Numbers of TUNEL+ cells with an apoptotic morphology (shrunken or fragmented nuclei) in SVZ did not differ between groups (2.9 ± 0.6 cells in TNF-R1−/−and 1.8 ± 0.5 cells in wild-type mice).
Hypothetically, the increased number of newly proliferated neuroblasts in the SVZ of TNF-R1−/−mice could be because of decreased migration. Tumor necrosis factor-α can induce migration of neutrophils (Oliveira et al, 2008) and smooth muscle cells (Rajesh et al, 2008). Moreover, administration of a TNF-α-converting enzyme proteolytic inhibitor to SVZ cultures reduced neuroblast migration (Katakowski et al, 2007). We did not find any BrdU + /Dcx+ cells in the injured striatum, rostral migratory stream, or olfactory bulb, probably because the time point was too early for them to have left SVZ. No differences in the density of Dcx + cells in rostral migratory stream were observed between wild-type and TNF-R1−/− mice, either ipsilateral (2.5 times 105 ± 4.9 times 104 cells/mm3 in wild-type and 3 times 105 ± 2.4 times 104 cells/mm3 in TNF-R1−/− mice) or contralateral to the MCAO (2.5 times 105 ± 3.7 times 104 cells/mm3 in wild-type and 2.6times105 ± 1.7 times 104 cells/mm3 in TNF-R1−/− mice). Similarly, we found no differences in the total numbers of Dcx+ cells in the striatum between wild-type and TNF-R1−/− mice ipsilateral (29 ± 0.7 cells in wild-type and 31 ± 1 cells in TNF-R1−/− mice) or contralateral to the ischemic damage (17 ± 0.7 cells in wild-type and 18 ± 0.6 cells in TNF-R1−/− mice). Also, the distance the new neuroblasts migrated into the damaged striatum did not differ between groups (data not shown). In line with more efficient neuroblast generation, we observed higher numbers of Dcx + cells in the SVZ ipsilateral to the damage in TNF-R1−/− than in wild-type mice (Supplementary Figure 1).
Deletion of TNF-R1 has been reported to ameliorate ischemic damage (Hallenbeck, 2002) and we have previously shown (Thored et al, 2006) that the extent of striatal injury influences the magnitude of the neurogenic response after stroke. We wanted to exclude the possibility that the increased SVZ cell proliferation in TNF-R1−/− mice was an indirect effect, caused by alterations in the size of the ischemic injury. In NeuN- and Fluoro-Jade- (data not shown) stained sections, we found no differences in striatal infarct volume between wild-type (15.1 ± 1.0 mm3; n = 6), TNF-α−/− (13.4 ± 1.0 mm3; n = 5), and TNF-R1−/− (14.4 ± 2.0 mm3; n = 8) mice after stroke, amounting to 79%, 80%, and 81% of the total volume, respectively (Figures 3D–3G).
We finally analyzed the effect of deletion of TNF-α on cell proliferation 1 week after MCAO. Also in this experiment, we observed a significant increase in the number of proliferating SVZ cells compared with intact animals using all three markers. However, no differences in the number of BrdU +, p-H3 +, and PCNA+ cells in SVZ, or the size of striatal injury, were detected between wild-type and TNF-α−/− mice (Figures 1E–1G and 3E and 3F).
Deletion of TNF-R1 Does Not Affect the Increased Progenitor Proliferation in the SVZ After SE
We then explored if the enhancement of cell proliferation caused by deletion of TNF-R1 was specific for stroke or also occurred in other pathologic situations with increased proliferation in SVZ. Animals were subjected to SE for 2 h and during this period showed partial or generalized convulsive behavior. We previously observed no differences between TNF-R1−/− and wild-type mice in any seizure parameter, that is, percentage of partial and generalized convulsions, or time to develop continuous ictal hippocampal EEG activity (Iosif et al, 2006).
We analyzed cell proliferation in the SVZ at 1 week after SE. The epileptic condition gave rise to significant increases (25% to 30%) in the number of proliferating BrdU +, p-H3 +, and PCNA + cells in both wild-type and TNF-R1−/− mice as compared with their electrode-implanted, nonstimulated controls (Figure 4). We found no differences in SVZ in any of the cell proliferation markers between the two groups of mice after SE (Figure 4).
Stroke but not SE Increases Microglia Numbers in the SVZ: No Effect of TNF-α or TNF-R1 Deletion
Because microglia is a major source of TNF-α, we explored if the numbers of these cells in SVZ are differentially regulated after stroke and SE, and influenced by deletion of TNF-R1 or its ligand. Tumor necrosis factor-α can induce microglia proliferation by means of TNF-R1 (Dopp et al, 1997) and effects of TNF-R1 deletion on SVZ cell proliferation could hypothetically be caused by changes in microglia numbers. As a marker for microglia, we used Iba1. No changes in SVZ Iba1 + cell numbers were observed in wild-type mice subjected to SE (Figure 5). In contrast, we found that MCAO gave rise to major increases in the numbers of Iba1 + cells bilaterally in the SVZ (Figure 5A). Numerous Iba1 + cells were detected not only in the SVZ but also in the striatum ipsilateral to MCAO, whereas high numbers of these cells on the contralateral side were found only in SVZ. Neither in intact mice nor after SE or MCAO did deletion of TNF-R1 or TNF-α cause any changes in the number of Iba1 + cells in the SVZ compared with wild type (Figure 5).
TNF-R1 is Expressed by Neural Stem/Progenitor Cells in SVZ and Upregulated Together with TNF-α After Stroke
To explore if the observed effect of TNF-R1 signaling on SVZ cell proliferation could be mediated directly on the progenitor cells themselves, we analyzed the presence of TNF-R1 using RT-PCR. The mRNA of TNF-R1 was expressed in SVZ tissue and in primary and expanded neurospheres (Figure 6A). We then sorted GFP+ and GFP- cells from the SVZ of nestin-GFP mice. In these animals, GFP is expressed under the nestin promoter and, therefore, labels progenitor cells in the SVZ (Mignone et al, 2004). We found expression of TNF-R1 in GFP + and interestingly also in GFP- cells (Figure 6A). Levels of TNF-α were very low or undetectable by RT-PCR under basal conditions.
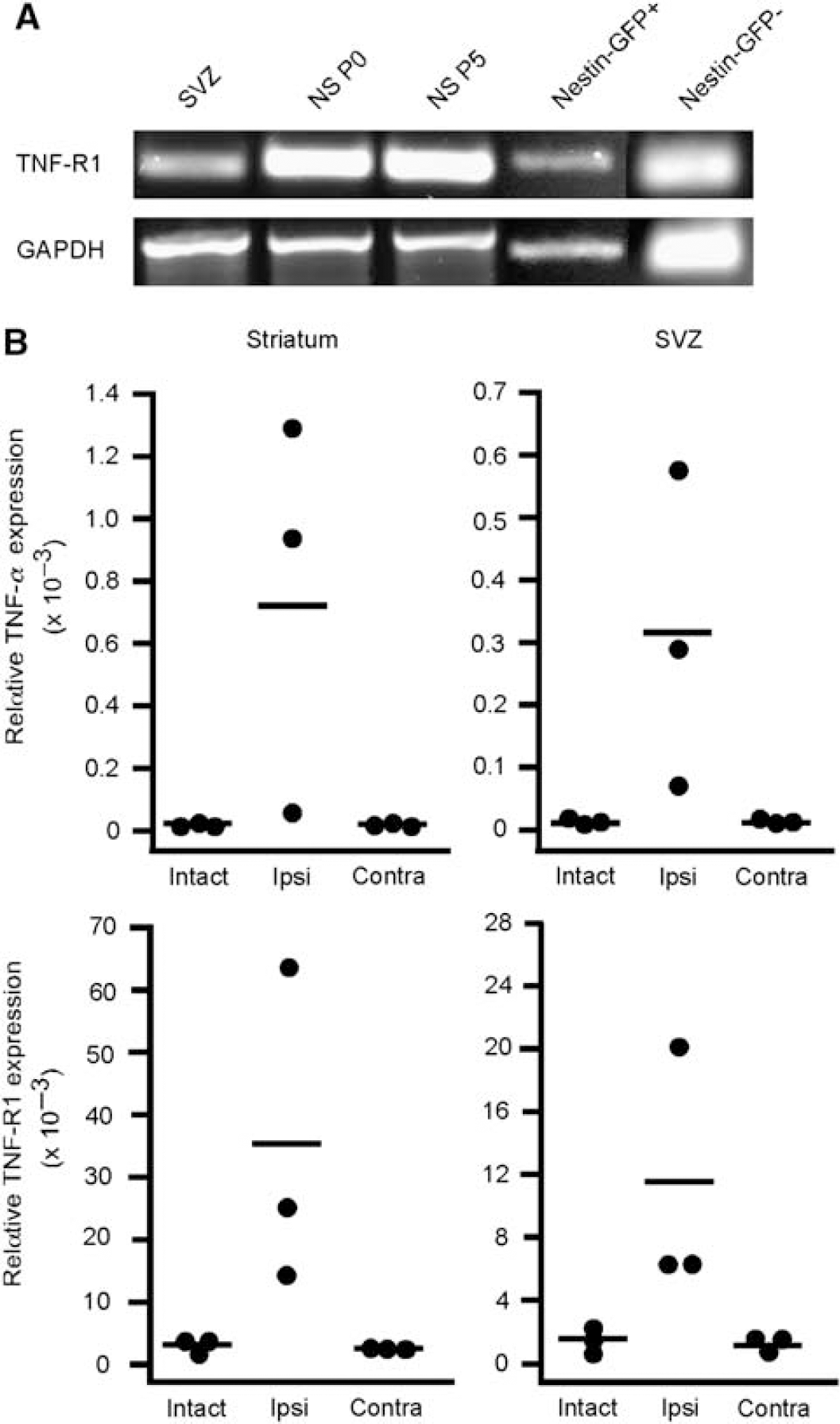
TNF receptor-1 is expressed in SVZ neural progenitor cells and upregulated together with TNF-α after stroke. (
Both TNF-α and TNF-R1 are upregulated in the stroke-damaged brain (Lambertsen et al, 2005) but whether any changes occur in SVZ is unknown. We subdissected SVZ and striatum 1 week after stroke and analyzed gene expression for TNF-α and TNF-R1 using quantitative real-time PCR. We found massive upregulation of TNF-α (296-fold) and TNF-R1 mRNA (8-fold) in the stroke-injured striatum, and also a 50- and 6-fold upregulation of TNF-α and TNF-R1 mRNA, respectively, in the tissue dissected from the ipsilateral SVZ (Figure 6B). Levels of expression of ligand and receptor in contralateral SVZ were not changed.
Tumor Necrosis Factor-α Decreases Proliferation of Neural Stem/Progenitor Cells Through TNF-R1 in SVZ Neurosphere Cultures
To further establish if deletion of TNF-R1 influences SVZ progenitor proliferation, we isolated cells from the SVZ of wild-type and TNF-R1−/− mice and used the neurosphere formation assay. In agreement with in vivo data, we found no differences between wild-type and TNF-R1−/− mice in numbers or size of primary neurospheres under basal conditions (Figure 7A). However, when recombinant mouse TNF-α was added, there was a 30% and 25% reduction in the numbers and size, respectively, of neurospheres formed from wild-type SVZ. In contrast, in cultures from TNF-R1−/− mice, addition of TNF-α had no effect on sphere numbers or size (Figure 7A). As signaling through TNF-R1 can induce cell death (Fontaine et al, 2002), we explored whether the effects of TNF-α addition were because of decreased survival. The lactate dehydrogenase test was used to assess general cell death (Figure 7B and Supplementary Figure 2) and TUNEL staining to show apoptosis (Figure 7C). However, we did not detect any differences between wild-type and TNF-R1−/− mice under basal conditions or after the addition of TNF-α to the cultures.
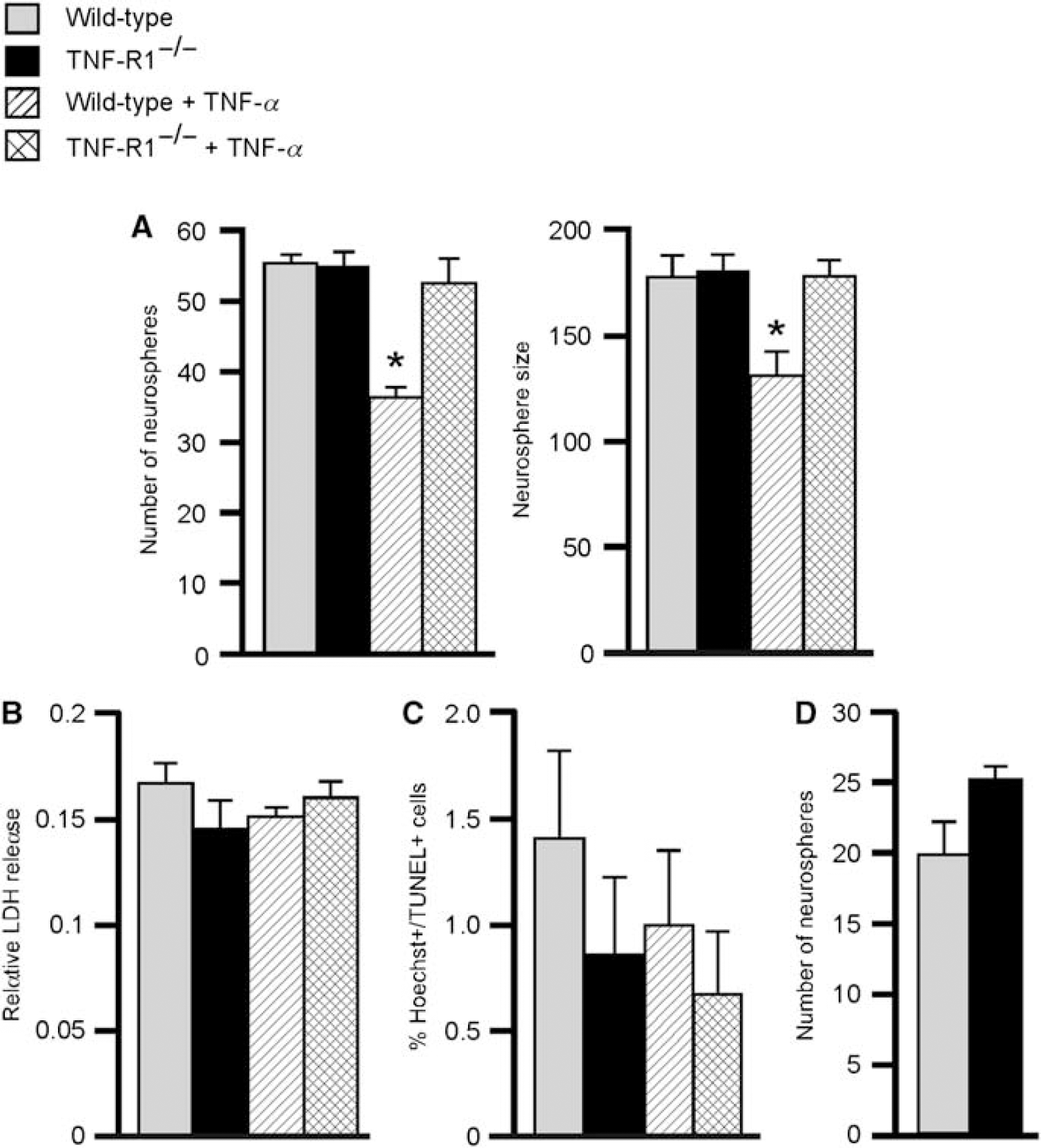
Tumor necrosis factor-α inhibits neural progenitor proliferation without affecting survival in vitro. (
We compared neurosphere formation from the SVZ of wild-type and TNF-R1−/− mice at 1 week after stroke, that is, at the time point when we had found upregulation of TNF-α and TNF-R1 gene expression. There was a trend (P = 0.08) toward increased number of neurospheres formed in the tissue from TNF-R1−/− mice (Figure 7D). The most likely explanation to this modest change is that the levels of endogenously produced TNF-α in the stem/progenitor cells in vitro were too low to significantly suppress neurosphere formation.
Discussion
Here, we show that mice with loss of TNF-R1 function respond to stroke with enhanced SVZ cell proliferation. In contrast, deletion of TNF-R1 did not influence basal cell proliferation in SVZ. However, we have previously found increased number of proliferating cells in the SGZ in intact TNF-R1−/− mice (Iosif et al, 2006), indicating that under basal conditions, TNF-R1 signaling acts to suppress progenitor proliferation in SGZ but not in SVZ.
Several lines of evidence indicate that the higher number of newly formed cells in the SVZ of TNF-R1−/− mice after stroke was because of increased proliferation of neural stem/progenitor cells. First, the number of proliferated BrdU + /Dcx + was higher in TNF-R1−/− as compared with wild-type mice. Second, deletion of TNF-R1 did not alter the elevated microglia numbers in the SVZ. Third, we found expression of TNF-R1 in the GFP + progenitor cells sorted from SVZ tissue of nestin-GFP mice. Because we also detected TNF-R1 expression in the GFP- fraction, the effect of TNF-R1 signaling on progenitor proliferation could also be indirect and mediated through other cells in the SVZ, such as microglia or astrocytes, which are known to express TNF-R1 (Dopp et al, 1997). Fourth, in the in vitro experiments, addition of TNF-α decreased neurosphere size and numbers through a TNF-R1-dependent mechanism without affecting survival. Taken together, our results provide the first evidence that TNF-R1 signaling mediates a suppressant effect on the proliferation of progenitor cells in the SVZ after stroke.
Previous work has shown a similar role of TNF-R1 signaling in other stem/progenitor cell systems. Activation of TNF-R1 inhibited proliferation of hematopoietic stem cells (Dybedal et al, 2001). Also, TNF-α suppressed the proliferation of TNF-R1-expressing neural progenitors derived from neonatal rat striatum (Ben-Hur et al, 2003). We have recently obtained evidence indicating that in SGZ, TNF-R1 is a negative regulator of progenitor proliferation both under physiologic conditions and after SE (Iosif et al, 2006). Seemingly in contrast to our present data, Katakowski et al (2007) observed that proliferation in the SVZ was promoted by the protease activity of TNF-α-converting enzyme. This discrepancy is most likely explained by the fact that besides being a convertase for TNF-α, TNF-α-converting enzyme activates many other substrates, for example, growth factors and cytokines, which could promote proliferation (Katakowski et al, 2007). The results presented here are also at variance with those of Widera et al (2006), who reported that TNF-α triggered proliferation of neural stem cells derived from the adult SVZ. However, Widera et al (2006) studied cultured cells derived from rats, whereas we have analyzed cell proliferation in the SVZ of mice both in vitro and in vivo. Also, we specifically investigated signaling through TNF-R1 in knockout mice, whereas Widera et al (2006) used human recombinant TNF-α, supposedly acting only through TNF-R1. Crossreactivity of human TNF-α on TNF-R2 or species differences in signaling through TNF receptors cannot be excluded.
In contrast to the effects of deletion of TNF-R1 on the SVZ response after stroke, we observed no changes in proliferation in mice with loss of TNF-α function. Similarly, basal proliferation in SGZ did not differ between TNF-α−/− and wild-type mice (RE Iosif et al, unpublished observations) even if there were alterations in mice with deletion of the receptors (Iosif et al, 2006). Also, retinal ischemia, which caused reduced and increased neuronal loss in TNF-R1−/− and TNF-R2−/− mice, respectively, did not induce any changes in TNF-α−/− mice (Fontaine et al, 2002). Taken together, these findings indicate the possible existence of a TNF-α-independent mechanism operating, for example, through the action of another ligand such as lymphotoxin α, which binds to TNF-R1 (Wajant et al, 2003). This mechanism could, hypothetically, be compensatory and a consequence of the ablation of TNF-α already during embryonic development.
Loss of TNF-R1 function had differential effects on SVZ cell proliferation following the two pathologic conditions stroke and SE. Although both insults gave rise to increased cell proliferation in SVZ, the dampening action of TNF-R1 on proliferation was revealed only after stroke. It is interesting to note that at the time of increased cell proliferation, that is, 1 week after stroke or SE, we detected high numbers of microglia in the SVZ of stroke-damaged brains, but not in mice subjected to SE. The elevated number of microglia in the SVZ, possibly associated with increased release of TNF-α, could attenuate the stroke-induced proliferative response through TNF-R1 signaling. It should be pointed out, though, that at 1 week after MCAO in wild-type animals, we detected elevated TNF-α and TNF-R1 mRNA levels only in SVZ tissue from the side ipsilateral to the damage. In contrast, the enhancement of progenitor proliferation in the SVZ of TNF-R1−/− mice was observed bilaterally. This discrepancy could indicate that the TNF-R1-mediated suppressant effect on progenitor proliferation is exerted by bilateral release of TNF-α or another ligand, possibly from microglia, already before the 1-week time point. The high TNF-α mRNA level in ipsilateral SVZ despite bilaterally increased microglia numbers suggests that the microglia in SVZ on the side of the damage at 1 week after stroke had adopted a more cytotoxic phenotype as compared with contralateral SVZ (Schwartz et al, 2006). The observed ipsilateral and contralateral effects on proliferation could also be because of elevated levels of TNF-α in serum and cerebrospinal fluid, which have been reported in patients after stroke (Zaremba and Losy, 2001) and would not be detected with gene expression analysis. Finally, it cannot be excluded that the elevated mRNA expression in ipsilateral SVZ samples was because of inclusion of some tissue from the damaged striatum, in which we found TNF-α and TNF-R1 mRNA levels to be much higher.
In this study, we have identified the first negative regulator of SVZ progenitor proliferation after stroke. Many compounds and treatments have been reported to increase SVZ cell proliferation after stroke (for references, see Lindvall and Kokaia, 2008). Much less is known about endogenous mechanisms regulating progenitor proliferation in the SVZ. Notch1 and its naturally occurring activator, Jagged1, are expressed in the adult SVZ (Stump et al, 2002) and administration of Notch ligands increases the number of proliferating cells in the SVZ after stroke (Androutsellis-Theotokis et al, 2006). Insulin-like growth factor-1 has also been proposed to be a mediator of the increased SVZ progenitor proliferation after stroke (Yan et al, 2006).
The present data provide evidence for a suppressant role of TNF-R1 signaling in progenitor proliferation during stroke-induced neurogenesis. TNF receptor-2 had no significant influence on cell proliferation in the SGZ (Iosif et al, 2006) and this receptor was, therefore, not studied here. However, TNF-α and its receptors most likely affect also other steps of neurogenesis after stroke. Consistent with a role for the survival of the new neurons, infusion of an antibody against TNF-α reduces the number of striatal neuroblasts generated after stroke, possibly by inhibiting a neuroprotective action of TNF-α mediated by TNF-R2 (Heldmann et al, 2005). Hypothetically, TNF-α could also influence the functional synaptic integration of the new neurons, for example, by modulating excitatory synaptic transmission (Pickering et al, 2005). Obviously, the actions of TNF-α and its receptors are complex and various consequences for stroke-induced neurogenesis could be envisaged. The present data suggest that suppression of TNF-R1 signaling might be a novel strategy to promote the proliferative response in the SVZ after stroke. However, it will be important to establish how such an approach will affect other steps of neurogenesis and the functional outcome after stroke.
Footnotes
Acknowledgements
We thank M Lundahl and U Sparrhult-Björk for technical assistance. This work was supported by the Swedish Research Council, Juvenile Diabetes Research Foundation, Swedish Diabetes Foundation, EU project LSHB-CT-2006-037526 (STEM-STROKE), and the Söderberg, Crafoord, Kock, and King Gustav V and Queen Victoria Foundations. The Lund Stem Cell Center is supported by a Center of Excellence grant in Life Sciences from the Swedish Foundation for Strategic Research.
None.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.