
Abstract
Transient focal ischemia is known to induce proliferation of neural progenitors in adult rodent brain. We presently report that doublecortin positive neuroblasts formed in the subventricular zone (SVZ) and the posterior peri-ventricle region migrate towards the cortical and striatal penumbra after transient middle cerebral artery occlusion (MCAO) in adult rodents. Cultured neural progenitor cells grafted into the non-infarcted area of the ipsilateral cortex migrated preferentially towards the infarct. As chemokines are known to induce cell migration, we investigated if monocyte chemoattractant protein-1 (MCP-1) has a role in post-ischemic neuroblast migration. Transient MCAO induced an increased expression of MCP-1 mRNA in the ipsilateral cortex and striatum. Immunostaining showed that the expression of MCP-1 was localized in the activated microglia and astrocytes present in the ischemic areas between days 1 and 3 of reperfusion. Furthermore, infusion of MCP-1 into the normal striatum induced neuroblast migration to the infusion site. The migrating neuroblasts expressed the MCP-1 receptor CCR2. In knockout mice that lacked either MCP-1 or its receptor CCR2, there was a significant decrease in the number of migrating neuroblasts from the ipsilateral SVZ to the ischemic striatum. These results show that MCP-1 is one of the factors that attract the migration of newly formed neuroblasts from neurogenic regions to the damaged regions of brain after focal ischemia.
Introduction
In the normal adult brain, neuroblasts derived from the neural progenitors in the subventricular zone (SVZ) migrate to the olfactory bulb via the rostral migratory stream, where they differentiate into interneurons (Alverez-Buylla and Garcia-Verdugo, 2002). However, the SVZ-derived neuroblasts migrate towards the ischemic striatum and cortex after focal ischemia in rodents (Arvidsson et al, 2002; Parent et al, 2002; Jin et al, 2003; Zhang et al, 2004). This suggests that factors produced in the ischemic regions might attract neuroblasts to migrate towards them. Focal ischemia induces microglial activation, astrocytosis, and leukocyte infiltration into the ischemic areas. All of these cells produce numerous factors including growth factors, cytokines and chemokines, which although not identified, were shown to play a role in neural progenitor proliferation, migration and differentiation (Aarum et al, 2003; Dempsey et al, 2003; Imitola et al, 2004; Robin et al, 2006; Yan et al, 2006).
Using GeneChip analysis, we recently identified that the expression of 15 diffusible factors, which have the potential to induce neural progenitor proliferation and migration, was significantly enhanced in the ipsilateral cortex after transient middle cerebral artery occlusion (MCAO) (Yan et al, 2006). Of these, monocyte chemoattractant protein-1 (MCP-1) has been shown to increase the migration of bone marrow stromal cells and neural progenitors in vitro (Wang et al, 2002; Widera et al, 2004). We currently report that MCP-1 mRNA expression increases significantly in the ipsilateral cortex and striatum between 6 h and 3 days after transient MCAO. To understand the functional significance of MCP-1 in ischemia-induced neurogenesis, we evaluated if cultured neural progenitors migrate towards MCP-1 in vitro and if the SVZ-derived neuroblasts migrate towards the striatum infused with MCP-1 in vivo. We also investigated whether MCP-1 and CCR2 (MCP-1 receptor) knockout mice show a curtailed neuroblast migration from the SVZ to the ischemic striatum.
Materials and methods
Animals
Animals were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services Publication number 86-23 (revised in 1986). The Research Animal Resources and Care Committee of the University of Wisconsin-Madison approved all the surgical procedures. In this study, adult spontaneous hypertensive rats (SHR, 270 to 300 g; Charles River, Wilmington, MA, USA) were used. CCR2- and MCP-1-deficient mice (20 to 25 g) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). These knockout mice were backcrossed eight times into a C57BL/6J background and were then bred as a homozygous MCP-1−/− or CCR2−/− line. Wild-type C57BL/6J mice (20 to 25 g, The Jackson Laboratory) were used as controls.
Focal Cerebral Ischemic Model
Focal ischemia was induced in rats and mice by intraluminal transient MCAO under halothane anesthesia as described in our previous studies (Dempsey et al, 2003; Yan et al, 2006; Kapadia et al, 2006). Briefly, the MCA was occluded for 1 h in both rats and mice with a monofilament nylon suture (3-0 for rats and 6-0 for mice). The occlusion and reperfusion were confirmed by measuring the regional cerebral blood flow with a laser-Doppler flowmeter as described earlier (Vemuganti et al, 2004). The sham-operated rats and mice were subjected to the same procedures but no intravascular filament occlusion was performed. Temporalis muscle and rectal temperatures were continuously monitored and maintained at 36.5°C to 37.0°C with a heating blanket and a heating lamp during the surgery. Mean arterial blood pressure was monitored and found to be 130 to 140 mm Hg in rats (this is the usual range for SHR) and 90 to 100 mm Hg in mice during MCAO and 10 min after reperfusion. The physiological parameters (PaCO2, PaO2, glucose, hemoglobin, Na+ and K+) were measured by iSTAT (Sensor Devices, Waukesha, WI, USA) before MCAO and at 10 min of reperfusion. All these parameters were at normal ranges as described previously (Yan et al, 2001).
To observe neuroblast migration after focal ischemia, 15 rats subjected to transient MCAO were killed at day 2, 4 and 6 of reperfusion (n = 5 per time point). Five sham-operated rats killed at day 2 after surgery served as control. Neuroblast migration was also studied in wild-type, MCP-1−/− and CCR2−/− mice. Mice subjected to transient MCAO were killed at day 5 of reperfusion (n = 6 of each strain). Sham-operated mice (n = 5 of each strain) killed at day 5 of sham surgery were used as controls. The brains were sectioned and subjected to anti-doublecortin (DCX) immunostaining. To evaluate the time course of MCP-1 protein expression after ischemia, rats subjected to transient MCAO were killed at day 1, 3 and 7 of reperfusion (n = 4 per group). A cohort of four sham-operated rats killed at day 1 of sham surgery served as control. MCP-1 protein expression was also examined in C57BL/6J mice. Sham operated wild-type mice at day 1 of sham surgery (n = 4), and ischemic wild type mice at day 1, 3 and 7 of reperfusion were killed (n = 4 per group). Brain sections were stained with anti-MCP-1 antibody. To identify the effect of MCP-1 and CCR2 knockout on progenitor proliferation, 15 sham-operated mice (n = 5 of wild-type, MCP-1−/− and CCR2−/−) at day 2 of sham surgery and 15 ischemic mice (n = 5 of wild-type, MCP-1−/− and CCR2−/−) at day 2 of reperfusion were injected with 5-bromo-2′-deoxyuridine (BrdU; 200 mg/kg, single injection, intraperitoneal, Sigma-Aldrich, St Louis, MO, USA) and killed at 24 h after injection. The brains were sectioned and stained with anti-BrdU antibody. This method labels dividing progenitors with less contamination of dividing daughter cells (Cameron and McKay, 2001). Infarct volumes were estimated in the wild-type, MCP-1−/− and CCR2−/− mice killed at day 5 of reperfusion after a 1 h MCAO (n = 6 of each strain) as described previously (Kapadia et al, 2006).
Real-Time PCR
To evaluate the time course of MCP-1 mRNA expression in the ischemic brain, rats were killed at 2, 6, 12 h, 1, 2, 3 and 5 days after transient MCAO (n = 4 per time point). Sham-operated rats (n = 4) were killed at day 1 of sham surgery. The cortices and striatum ipsilateral to MCAO or sham surgery were dissected (supplementary Figure A). The RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Total RNA (1 μg) from each sample was reverse transcribed with oligo(dT)15 and random hexamer primers using M-MuLV reverse transcriptase (Life Technologies, Rockville, MD, USA). Real-time PCR was conducted as described earlier (Vemuganti et al, 2004). Ten nanograms of cDNA and MCP-1 primers (5′-3′, CCA GAA ACC AGC CAA CTC TCA and GGC AGC AAC TGT GAA CAA CAG) were added to SYBR Green PCR Master Mix (SYBR Green I Dye, AmpliTaq® DNA polymerase, dNTPs with dUTP and optimal buffer components; Applied Biosystems, Foster City, CA, USA) and subjected to PCR amplification (one cycle at 50°C for 2 min, one cycle at 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 1 min) in a TaqMan 5700 Sequence Detection System (Applied Biosystems). Real-time PCR was conducted three times in duplicate using each of the RNA samples. The amplified transcripts were quantified with the comparative CT method using 18S rRNA as the internal control (http://docs.appliedbiosystems.com/pebiodocs/04303859.pdf). In previous studies, we did not observe significant change in 18S rRNA expression after transient focal ischemia (Vemuganti et al, 2004; Yan et al, 2006).
Neural Progenitor Culture
Adult, male SHR and transgenic mice expressing an enhanced green fluorescent protein (GFP) under the control of a chicken β-actin promoter and cytomegalovirus enhancer (The Jackson Laboratory) were deeply anesthetized with halothane. The brain was removed and put in a cold Dulbecco's phosphate buffered saline with 4.5 g/L glucose (DPBS/glu). The ependymal zones (including subependymal and subventricular zones from the lateral wall of the lateral ventricle) were dissected under a microscope, and then transferred to fresh DPBS/glu to rinse off blood. Tissues were then minced in cold Hank's balanced salt solution without Mg2+/Ca2+ (HBSS; Invitrogen), and treated with 0.01% papain (Worthington Biochemical Corporation, Lakewood, NJ, USA), 0.1% dispase II (Boehringer Mannheim, Indianapolis, IN, USA), 0.01% DNase I (Worthington Biochemical Corporation) and 12.4 mmol/L MgSO4 in HBSS at 37°C for 30 to 40 min. The cell suspension was triturated every 10 min, centrifuged, and the pellet was rinsed with Dulbecco's-modified eagle medium (DMEM)/F12 (1:1, Invitrogen). The cells were suspended in neural basal medium (Invitrogen) with B27 (without retinoic acid, Invitrogen), 2 mmol/L glutamine, 0.1 g/mL penicillin/streptomycin (Invitrogen), 2 μg/mL heparin (Sigma-Aldrich), 20 ng/mL FGF-2 (R&D Systems, Minneapolis, MN, USA), and seeded in T-25 culture flasks. The cultures were maintained at 37°C in an incubator with 5% CO2. Neurospheres formed within 5 to 7 days of suspension in culture. Neural progenitors were passaged once a week by dissociating neurospheres into single cells with accutase. The cells were maintained and used for experiments within 15 passages.
Immunohistochemistry
Animals were deeply anesthetized with halothane, and then transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4). Brains were removed, post-fixed in 4% paraformaldehyde overnight at 4°C, cryoprotected with 30% sucrose in PBS and sectioned coronally (30 μm). The free-floating method was used for both the avidin—biotin—peroxidase labeling and fluorescence staining. For avidin—biotin—peroxidase labeling, sections were rinsed with tris-buffered saline (TBS, pH 7.4), treated with 0.3% H2O2 (v/v) for 10 min, and denatured in 2N HCl for 1 h at 37°C (only for BrdU staining). After washing with TBS, the sections were incubated with a blocking solution (3% normal donkey serum, 0.1% Triton X-100 in TBS) for 30 min and probed with the desired primary antibody overnight at 4°C. After rinsing in TBS, sections were incubated for 1 h in biotinylated secondary antibody (1:200, Vector Laboratories, Burlingame, CA, USA). Sections were then washed three times in TBS and incubated for 45 min in avidin-peroxidase complex (1:100, Vector Laboratories). The color reaction was developed with a DAB kit (Vector Laboratories). For fluorescence double labeling, sections were incubated with blocking solution for 30 min, followed with incubation of primary antibodies overnight at 4°C. After rinsing in TBS, the sections were incubated with secondary antibodies for 1 h. The after primary antibodies were used: biotin conjugated mouse anti-BrdU (1:50, Molecular Probes, Eugene, OR, USA), rabbit anti-glial fibrillary acidic protein (GFAP, 1:2000, Dako, Carpinteria, CA, USA), rabbit anti-DCX (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-OX-42 (1:100, BD Biosciences, San Jose, CA, USA), goat anti-MCP-1 (1:20, R&D Systems), goat anti-CCR2 (1:100, Santa Cruz Biotechnology). Alexa Fluor 488 (green) and 594 (red) conjugated secondary antibodies (1:200, Molecular Probes) were used according to the combination of primary antibodies. Negative controls were performed by omitting primary or secondary antibodies.
Transplantation
The in vitro expanded SVZ-derived neurospheres from GFP expressing mice were dissociated into single cells by accutase, and approximately 5 × 105 cells in 3 μL culture medium were injected with a Hamilton syringe into the non-infarcted area of ipsilateral cortex (coordinates: anterior—posterior -0.3 mm; medial—lateral 2 mm; dorsal—ventral 2 mm based on the rat brain atlas of Paxinos and Watson, 1998) of ischemic rats at day 1 of reperfusion (n = 8). The same amount of cells was grafted into sham-operated rats as a control (n = 6). This site was chosen for cell transplantation because it allows us to determine whether the transplanted cells migrate preferentially towards the lesion area. To prevent graft rejection, rats were immunosuppressed by injecting cyclosporine once a day (10 mg/kg, intraperitoneal). All rats were killed at 2 weeks after transplantation.
In Vitro Migration Assay
The method of migration assay was modified from a growth cone turning assay (Lohof et al, 1992), which showed that a gradient near the growth cone was produced by a steady ejection of chemical solution from a micropipette against a perfusion flow of chemical solution across the entire culture. Three neurospheres derived from the SVZ of SHR were plated separately on a laminin-coated six-well plate. After the neurospheres attached to the plate, 3 mL of culture medium was added to each well. MCP-1 (R&D Systems) dissolved in neural basal medium with 0.1% bovine serum albumin (BSA) at 5, 50, and 500 ng/mL was pumped into the culture medium near the wall of the culture plate (approximately 0.5 to 1 cm away from the attached neurospheres) with an osmotic minipump (Model 1003D, DURECT Co., Cupertino, CA, USA) at the rate of 1 μL/h. BSA solution alone was pumped as a control. The pump was immersed in normal saline in the adjacent well of the culture plate (Figure 5D). After 3 days of incubation, the plates were stained with Hoechst (Sigma-Aldrich). The distance of cell migration out of the attached neurosphere towards the pump was measured (Figure 5A and 5C) and compared between groups. The data were collected from 10 to 12 neurospheres for each treatment group.
MCP-1 Infusion
MCP-1 was dissolved in artificial cerebrospinal fluid (aCSF; 119 mmol/L NaCl, 3.1 mmol/L KCl, 1.2 mmol/L CaCl2, 1 mmol/L MgSO4, 0.50 mmol/L KH2PO4, 25 mmol/L NaHCO3, 5 mmol/L
Cell Counting
DCX positive cells in the ipsilateral striatum or BrdU positive cells in the ipsilateral SVZ of each mouse were counted using an inverted microscope (Olympus America Inc, Melville, NY, USA) attached to a digital imaging system (Spot Camera, Diagnostic Instruments Inc., Sterling Heights, MI, USA) as described earlier (Dempsey et al, 2003). Sections (30 μm) at 180 μm intervals that cover the striatum and SVZ (+ 1.1 to -0.2 mm from bregma as per the Mouse Brain Atlas, Paxinos and Franklin, 1997) were used for each animal (seven sections from each mouse). The number of DCX positive cells in the striatum of seven sections was averaged in each animal and given as the mean ± s.d. For the counting of BrdU cells in the SVZ, each imaged SVZ was traced using the NIH ImageJ software (written by Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij). The traced area was measured and the total numbers of BrdU positive cells in the traced area were counted. For the counting of DCX positive cells in the BSA and MCP-1 infused striatum, four sections (90 μm interval) around the needle track from each rat were used. Four sections at a similar position were also selected from each of the sham-operated rats. The data were presented as total cells per mm2 ± s.d. The statistical analysis was conducted by one-way ANOVA followed by Bonferroni/Dunn post-test.
Results
Neuroblast Migration Induced by Focal Ischemia
In this study, DCX (a protein expressed in migrating and differentiating neurons) was used as a marker to examine neuroblast migration after focal ischemia (Francis et al, 1999). In the sham-operated rats, DCX positive cells were only observed in the SVZ (Figure 1A). At day 2 of reperfusion after transient MCAO, several DCX positive cells were observed in the penumbra of the striatum (Figure 1B), indicating the lateral migration of these cells from the SVZ, escaping the rostral migratory stream. The number of DCX positive cells in the penumbra of the striatum and the distance of cell migration further increased at day 4 and 6 of reperfusion (Figure 1C and Figure 1D). Compared with the sham brain (Figure 1A), the number of GFAP positive astrocytes also increased in the ischemic penumbra of the striatum after MCAO (Figure 1B–Figure 1D), indicating post-ischemic gliosis. The migrating DCX positive cells formed a chain along the GFAP positive astrocytes (inset in Figure 1C), suggesting that activated astrocytes might guide the migration of the SVZ-derived neuroblasts towards the injured area. In addition to striatal migration, the DCX positive cells were also observed to migrate towards the ischemic cortex from the ipsilateral SVZ (Figure 1E) and the posterior peri-ventricle (PPV, Figure 1F).
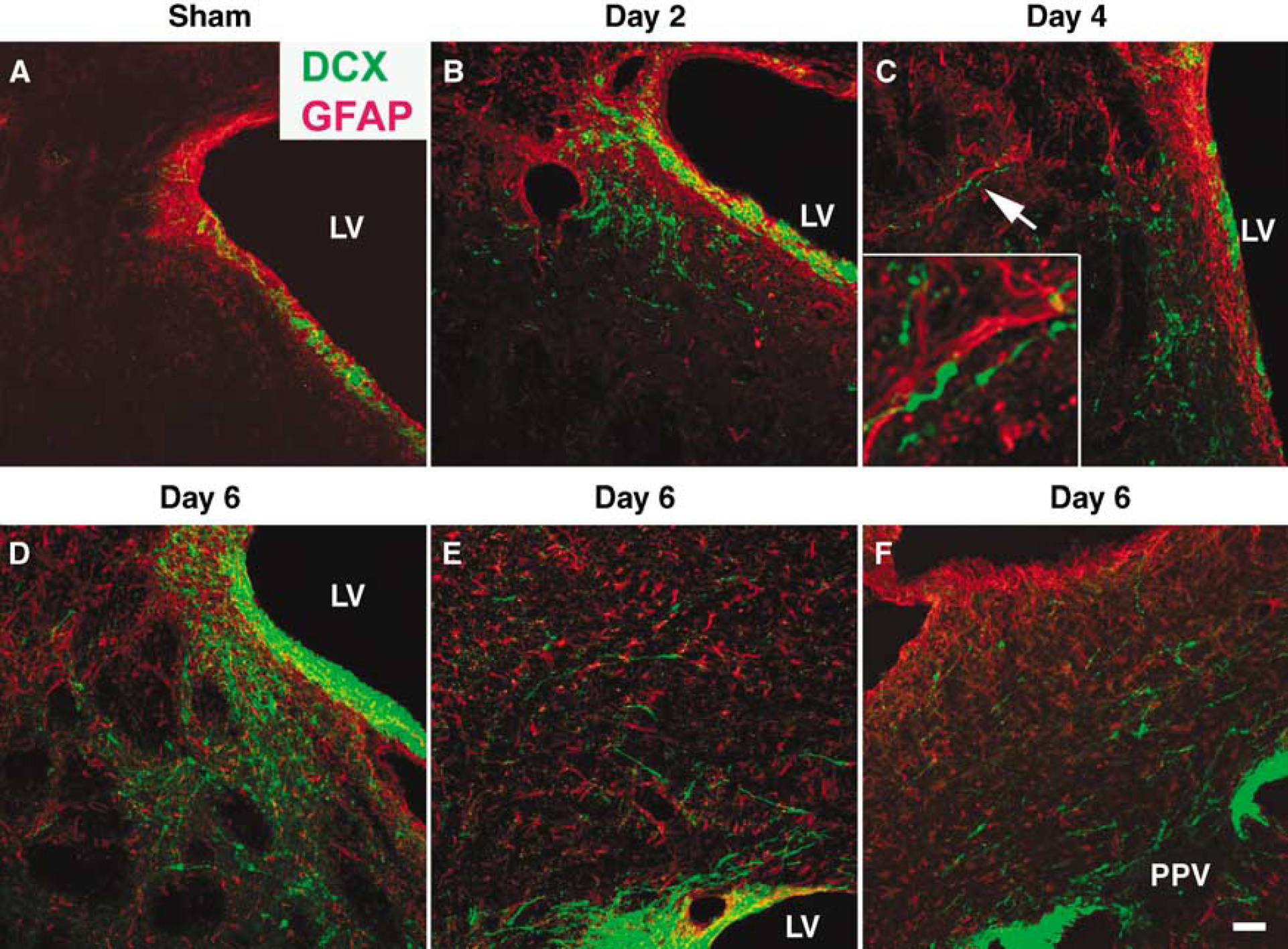
Focal ischemia-induced neuroblast migration from the ipsilateral SVZ and PPV to the injured areas in rat brain. DCX positive cells, which were restricted to the SVZ of the sham brain (
Transplanted Neural Progenitors Migrated towards the Infracted Area
As shown above, neuroblasts from the SVZ and PPV migrated to the injured regions after focal ischemia. This led us to hypothesize that the factors formed in the ischemic region attract the newborn neuroblasts to migrate towards it. In order to verify this hypothesis, in vitro expanded neural progenitors isolated from the SVZ of GFP transgenic mice were transplanted into the cortex of sham-operated rats or the non-infarcted area of the ipsilateral cortex of ischemic rats. In the sham rats (six of six), the grafted GFP expressing cells were observed to survive at the site of injection and migrated a short distance around the needle track into the adjacent cortex by 14 days (Figure 2A). In the ischemic rats (eight of eight), the cells grafted at day 1 of reperfusion migrated along the subcortical white matter from the injection site towards the infarcted area (Figure 2B). The cells migrated to the ischemic region (six of eight) showed a differentiated morphology with long processes (Figure 2C). Most of GFP expressing cell that migrated into the white matter were DCX positive (Figure 2D–Figure 2F).
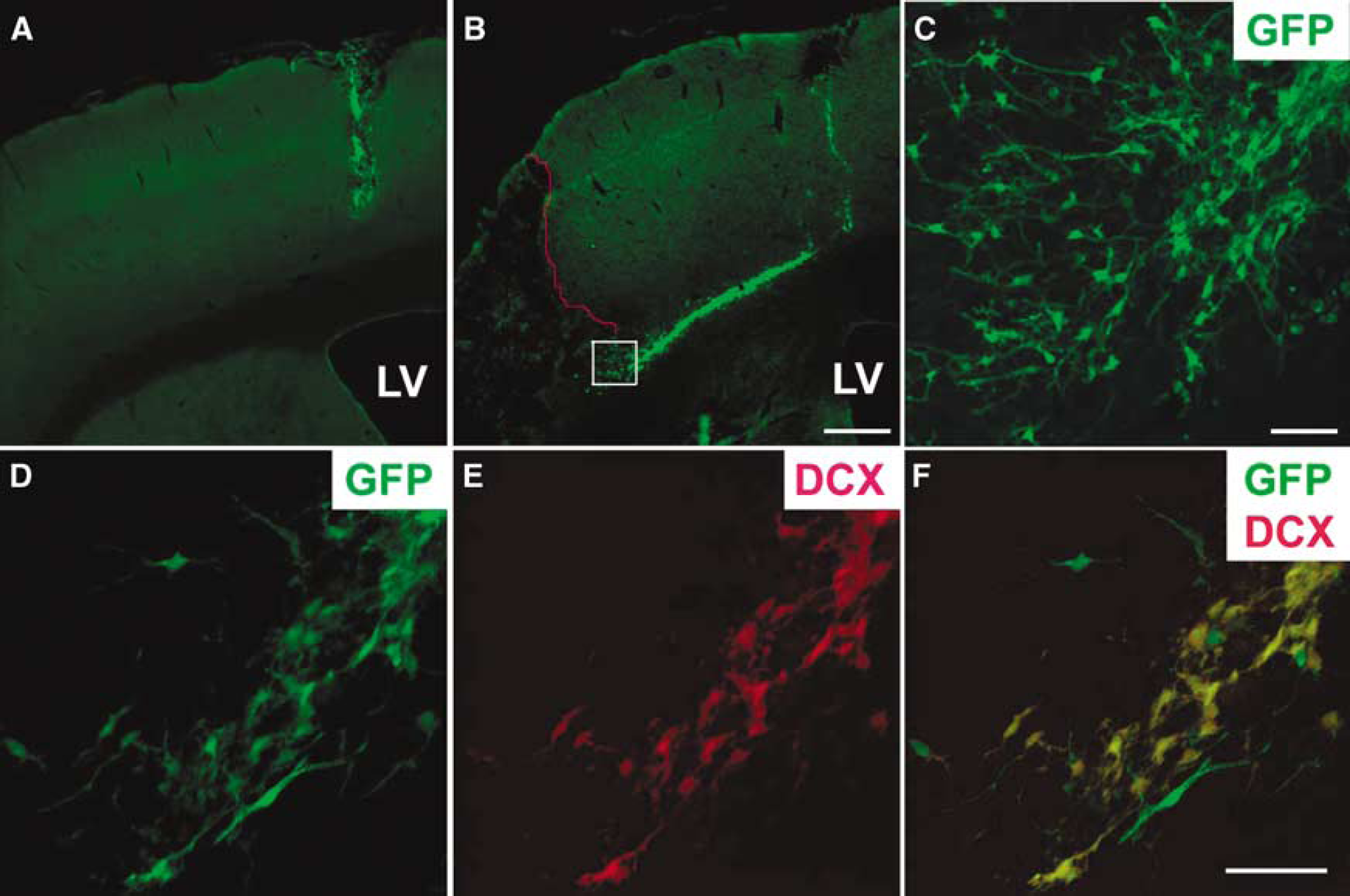
Ischemia-induced migration of neural progenitors transplanted into the non-infarcted area of the ipsilateral rat cortex. GFP expressing mouse neural progenitors were transplanted at day 1 of reperfusion and animals were killed at day 14 of transplantation. The grafted cells, which remained at the site of the graft in the sham cortex (
Increased MCP-1 Expression in the Ischemic Area
Using real-time PCR analysis, we estimated the MCP-1 mRNA expression in the ipsilateral cortex and striatum of rats subjected to transient MCAO (Supplementary Figure A). In both regions, there was a significant upregulation of MCP-1 mRNA levels (3.3- to 16.7-fold, P < 0.05) between 6 h to 3 days of reperfusion after transient MCAO as compared with sham control (Figure 3). The expression of MCP-1 mRNA returned to basal levels by 5 days of reperfusion (Figure 3). In the sham-operated rat brain, MCP-1 immunostaining was below the level of detection in the striatum (Figure 4A), the subcortical white matter (Figure 4B) and the cortex (Figure 4C). After focal ischemia, many MCP-1 positive cells were observed in the ipsilateral striatum (Figures 4D and Figure 4G), the subcortical white matter (Figure 4E and Figure 4H) and the cortex (Figure 4F and Figure 4I) at days 1 and 3 of reperfusion. Many of these MCP-1 positive cells expressed GFAP, typically in the ischemic white matter (Figure 4E and Figure 4H). The remaining MCP-1 positive cells in the ipsilateral striatum and cortex co-expressed the activated microglial maker OX-42 (Supplementary Figures B—D). These results suggest that astrocytes and activated microglia are the source of MCP-1 in the ischemic brain. The MCP-1 protein expression returned to sham levels in the ipsilateral striatum, subcortical white matter and cortex by day 7 of reperfusion (Figure 4J–Figure 4L).
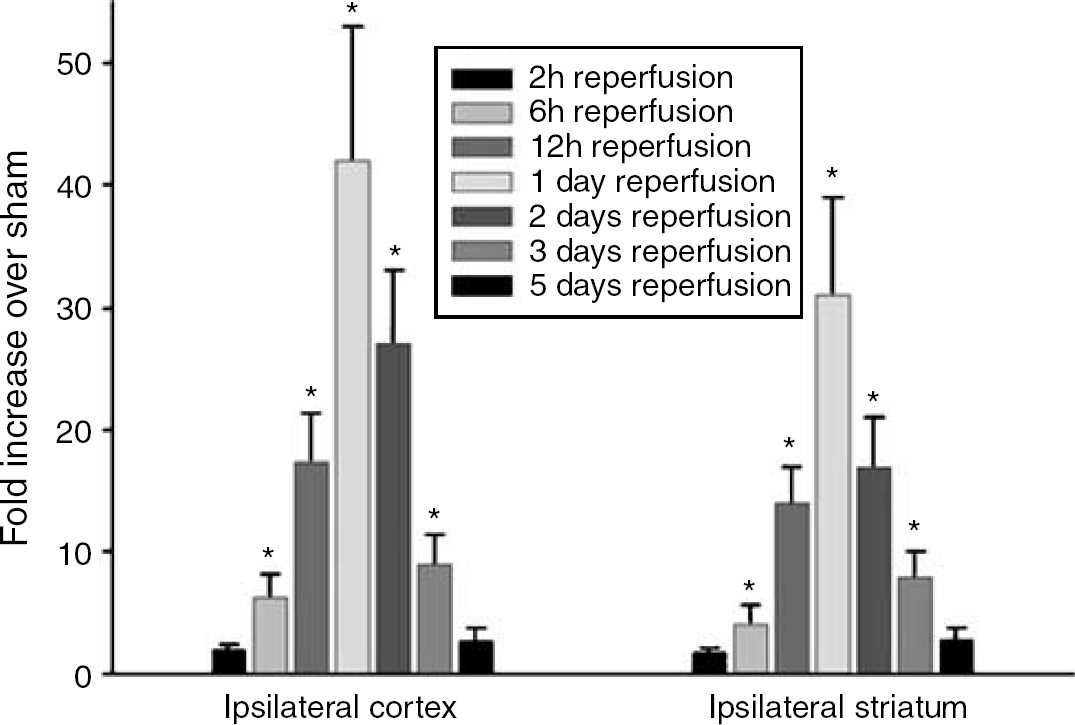
Upregulated MCP-1 mRNA expression in ischemic rat brain. Real time PCR showed that MCP-1 mRNA expression in the both ipsilaretal cortex and striatum increased between 6 h and 3 days, and returned to sham levels at 5 days after transient MCAO. Values in the histograms are mean ± s.d. *P < 0.05 versus sham.
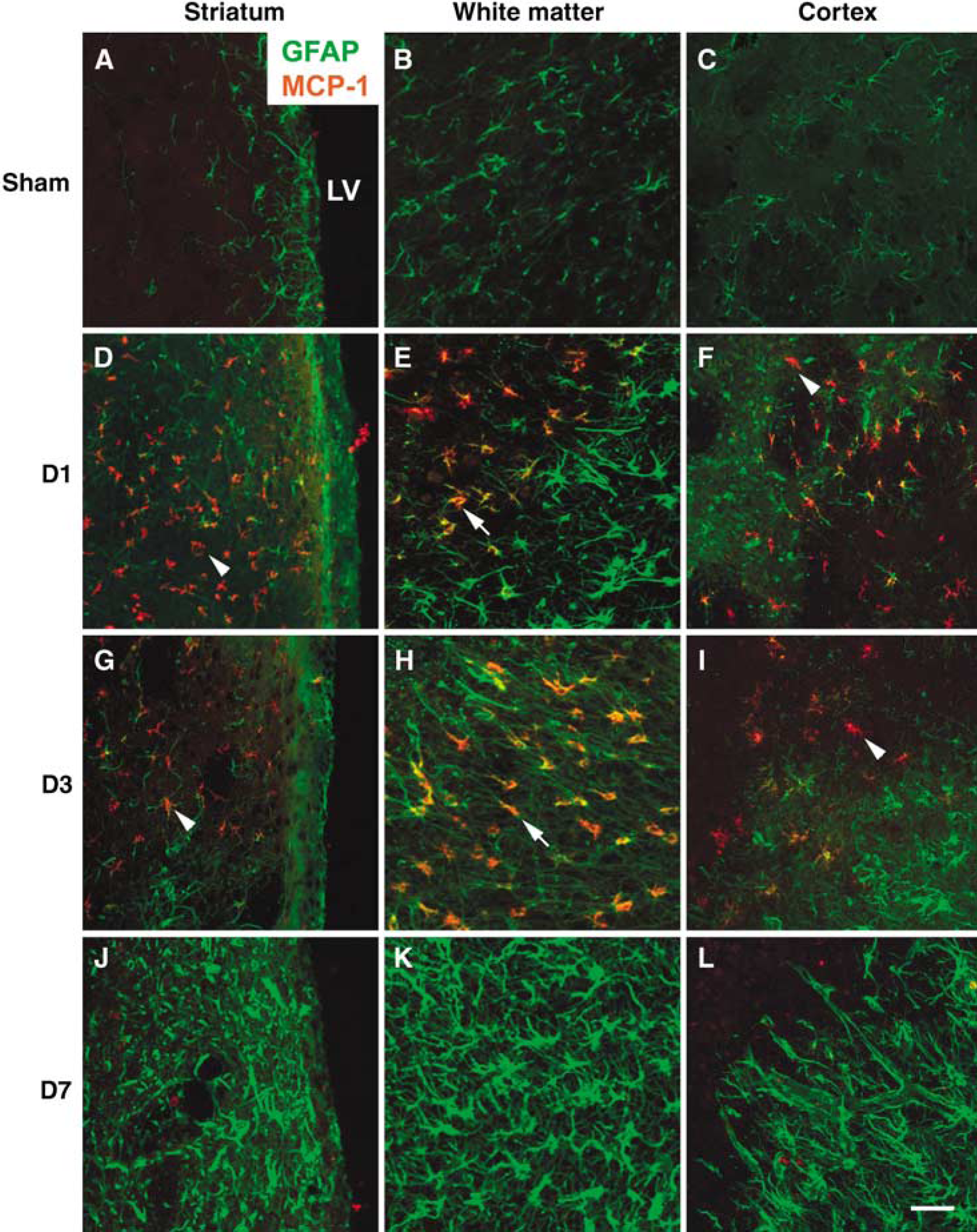
MCP-1 protein expression in rat brain after transient MCAO. The schematic of a brain section indicating where the images were taken was shown in supplementary Figure A. MCP-1 expression, which was not detectable in the sham striatum (
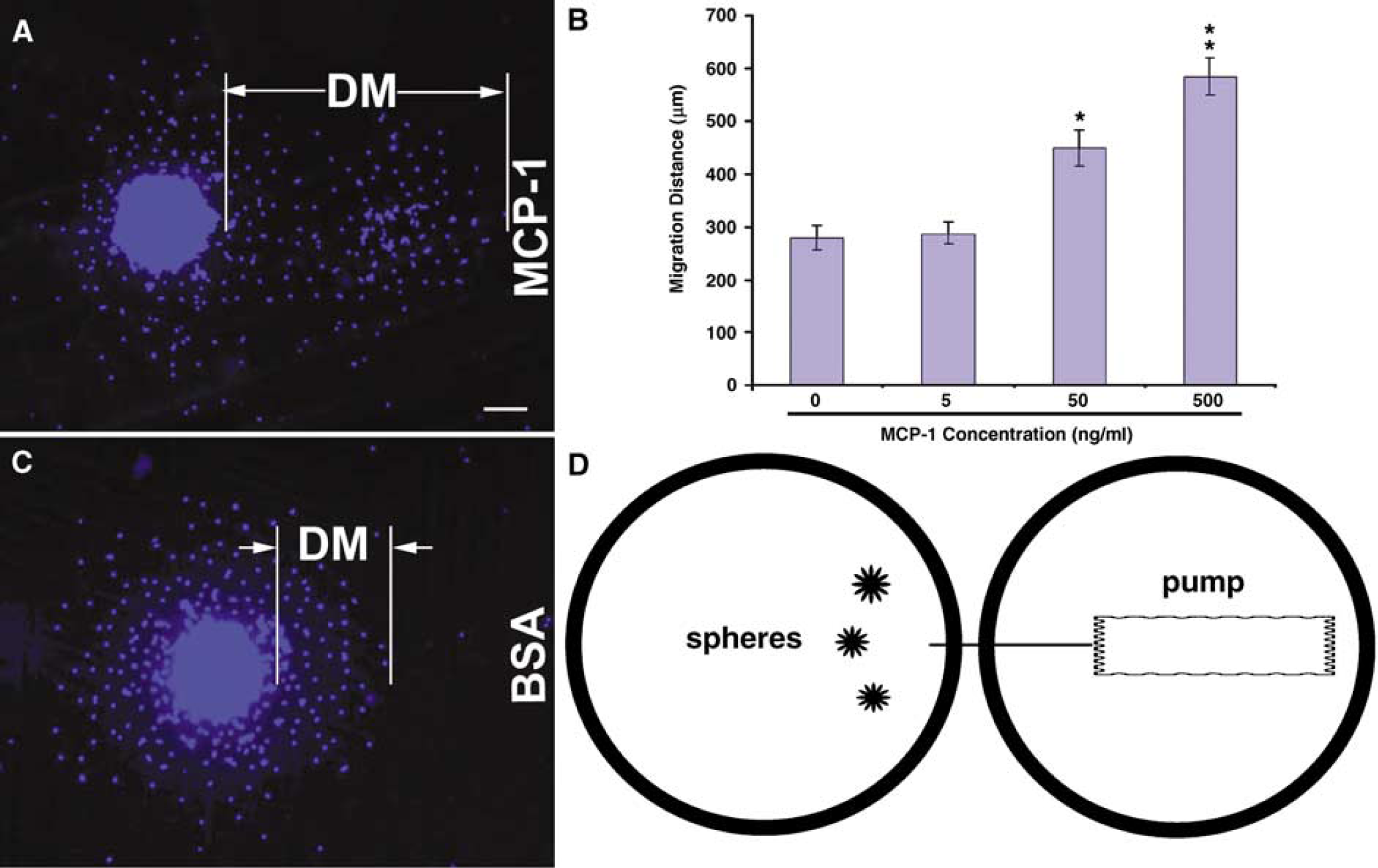
MCP-1 induced neural progenitor migration in vitro. (
Induction of Progenitor Migration by MCP-1 In Vitro and In Vivo
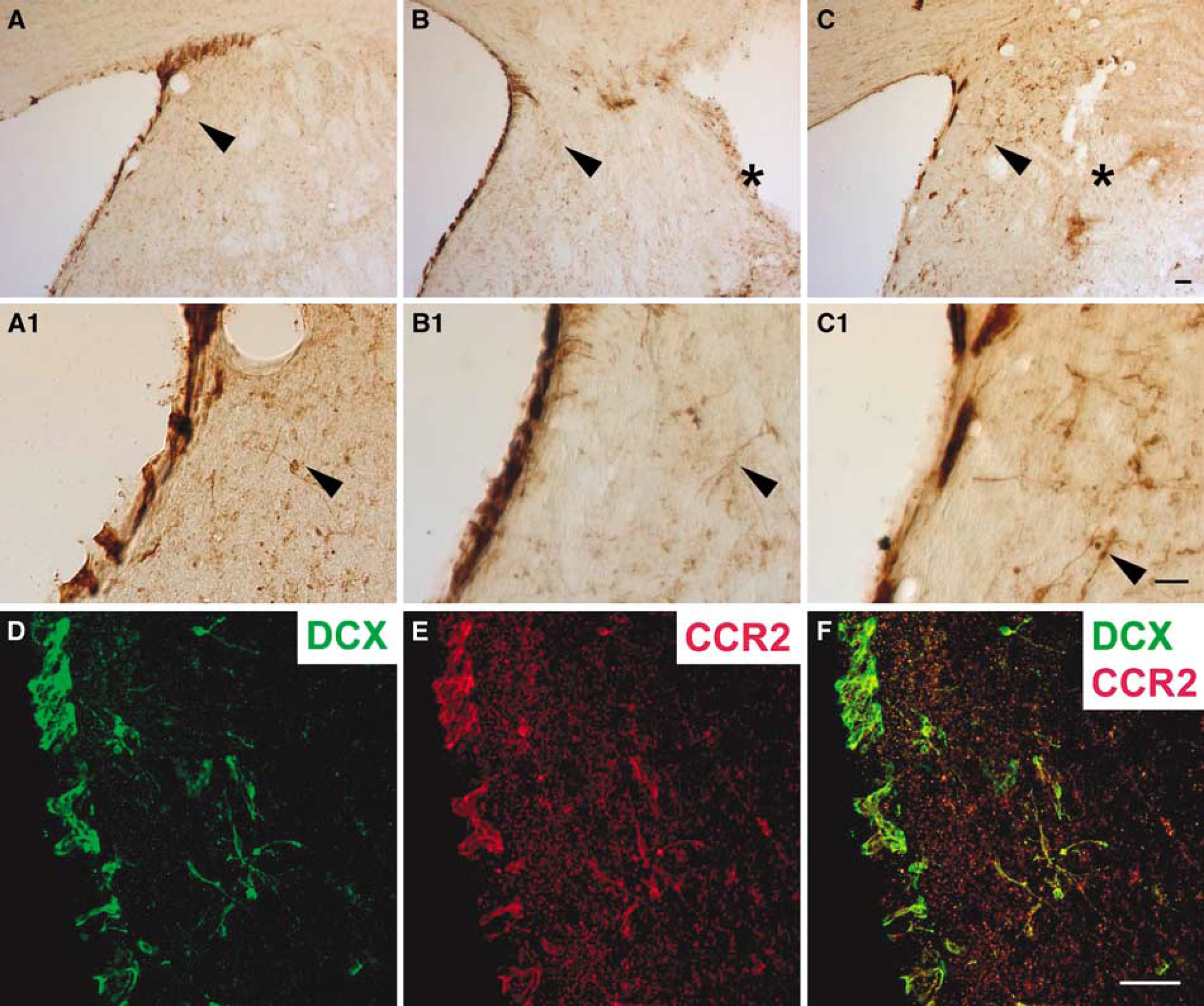
When MCP-1 was infused for 3 days to one edge of the culture plate containing neurospheres, the neural progenitors were observed to migrate significantly towards the site of infusion (Figure 5A). The distance of progenitor migration towards the site of infusion was dose-dependently increased by MCP-1 (Figure 5B), whereas the progenitors migrated equal distances in all directions when neurosphere cultures were treated with BSA (control) for 3 days (Figure 5C). Next, we examined if neuroblasts from the SVZ of normal rats would migrate towards MCP-1 infused in vivo. In sham-operated rats, few DCX positive cells were observed in the striatum close to the SVZ (Figure 6A and A1, 5.3 + 1.7 cells/section). Infusion of BSA into the striatum did not induce obvious neuroblast migration from the SVZ towards the site of infusion (Figure 6B and B1, 6.2 + 2.1 cells/section). However, many DCX positive cells were observed to migrate from the SVZ towards the site of infusion in the MCP-1-infused rat striatum (Figure 6C and C1, 48.3 ± 13.1 cells/section, P < 0.01 versus sham or BSA infusion). The DCX positive progenitors in the SVZ and those migrating to the striatum induced by MCP-1 infusion expressed the MCP-1 receptor CCR2 (Figures 6D–Figure 6F).
MCP-1 induced neural progenitor migration in vivo. Few DCX positive cells were observed in the striatum (arrowhead) of the sham (
MCP-1 and CCR2 Knockout Mice Showed Decreased Post-Ischemic Neuroblast Migration
We examined whether ablation of MCP-1 or its receptor CCR2 diminishes ischemia-induced neuroblast migration from the SVZ to the striatum. In the wild-type mouse brain, MCP-1 was expressed in astrocytes and activated microglia between days 1 and 3 of reperfusion after transient MCAO (data not shown). Thus, the pattern and timing of postischemic MCP-1 expression were similar between rat and mouse. In the brains of sham-operated wild-type mice, DCX positive cells were observed in the SVZ and almost no DCX positive cells were found in the adjacent striatum (Figure 7A). The DCX staining pattern in the brains of sham MCP-1 or CCR2 knockout mice was similar to that observed in the sham wild-type mice (data not shown). At day 5 of reperfusion, many DCX positive neuroblasts migrated into the ipsilateral striatum in the wild-type mice subjected to transient MCAO (Figure 7B). However, the number of DCX positive cells in the ipsilateral striatum was significantly less at day 5 of reperfusion in the MCP-1 knockout mice (Figure 7C) and the CCR2 knockout mice (Figure 7D) subjected to transient MCAO. Cell counting showed a threefold decrease of DCX positive cells in the ipsilateral striatum of MCP-1 and CCR2 knockout mice compared with the wild-type mice subjected to transient MCAO (Figure 7E).
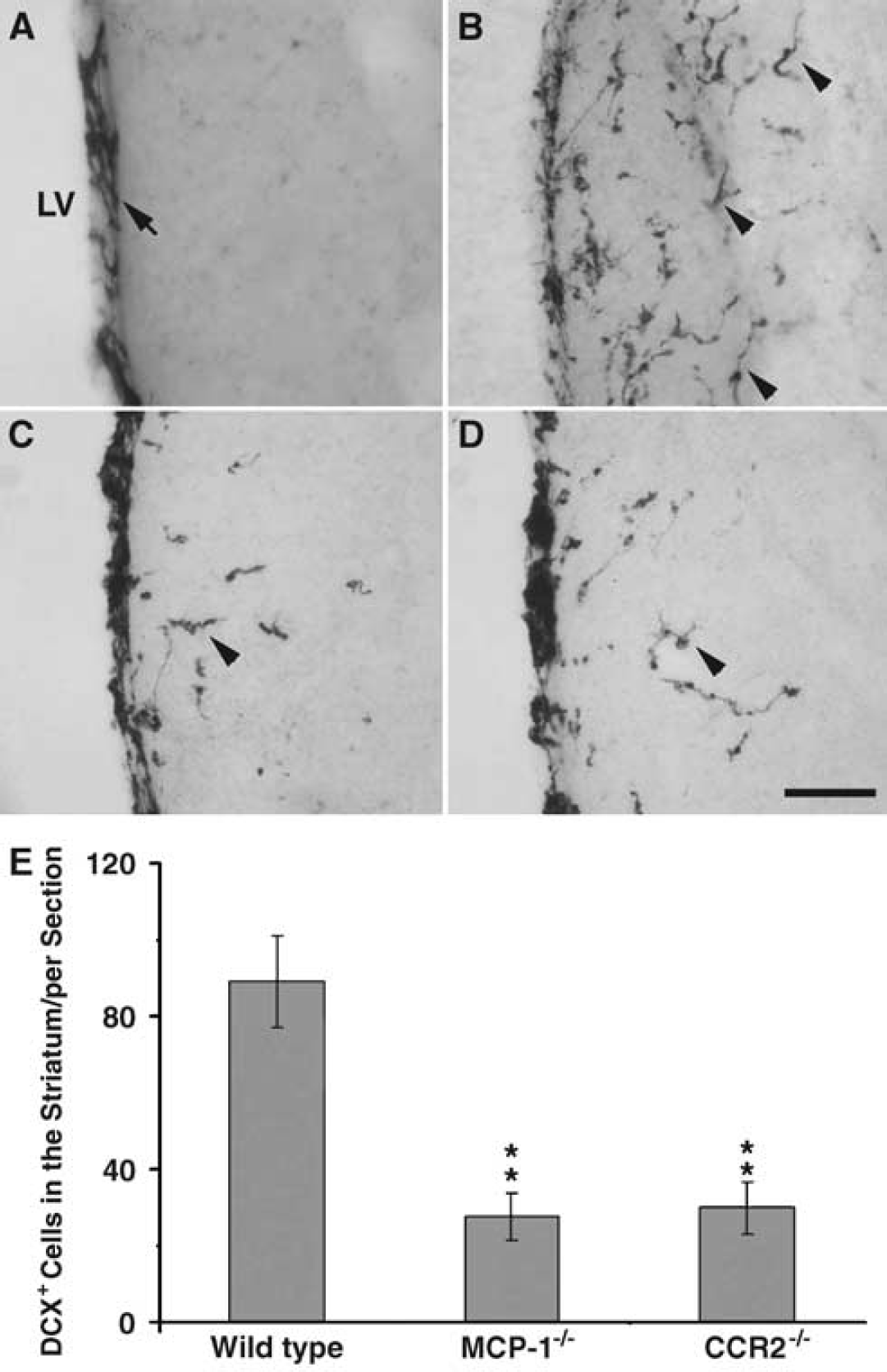
Ablation of MCP-1 or CCR2 decreased ischemia-induced neuroblast migration in mice. DCX positive cells, which were restricted to the SVZ (arrow) of sham wild-type mice (
Deficiency of MCP-1 or CCR2 did not Affect Neural Progenitor Proliferation
The reduction of DCX positive cells in the ipsilateral striatum of MCP-1 and CCR2 knockout mice might be because reduced migration or decreased progenitor proliferation in the SVZ. To examine progenitor proliferation, sham and ischemic mice were injected with BrdU once at day 2 of surgery and killed at 24 h later. In the sham-operated wild-type (Figure 8A), MCP-1 (Figure 8B), and CCR2 knockout mice (Figure 8C), BrdU positive cells were observed to be restricted to the SVZ. Cell quantification showed that there was no difference in the number of BrdU positive cells in the SVZ among these three types of mice (Figure 8G). Compared with the respective sham-operated mice, the number of BrdU positive cells in the ipsilateral SVZ increased significantly (by 2.7- to 3.2-fold) in the ischemic wild-type (Figure 8D), MCP-1 (Figure 8E), and CCR2 knockout mice (Figure 8F). However, cell quantification showed no difference in the number of BrdU positive cells in the ipsilateral SVZ between wild-type, MCP-1, and CCR2 knockout mice after ischemia in comparison to their respective sham controls (Figure 8G). The infarct volumes of wild-type, MCP-1, and CCR2 knockout mice were 24.0 ± 5.4, 19.8 ± 6.7 and 20.1 ± 7.1 mm3, respectively. There was no statistically significant difference between the three groups. These results suggest that ablation of MCP-1 or CCR2 does not affect progenitor proliferation in the SVZ, but reduces ischemia-induced neuroblast migration to the ischemic region.
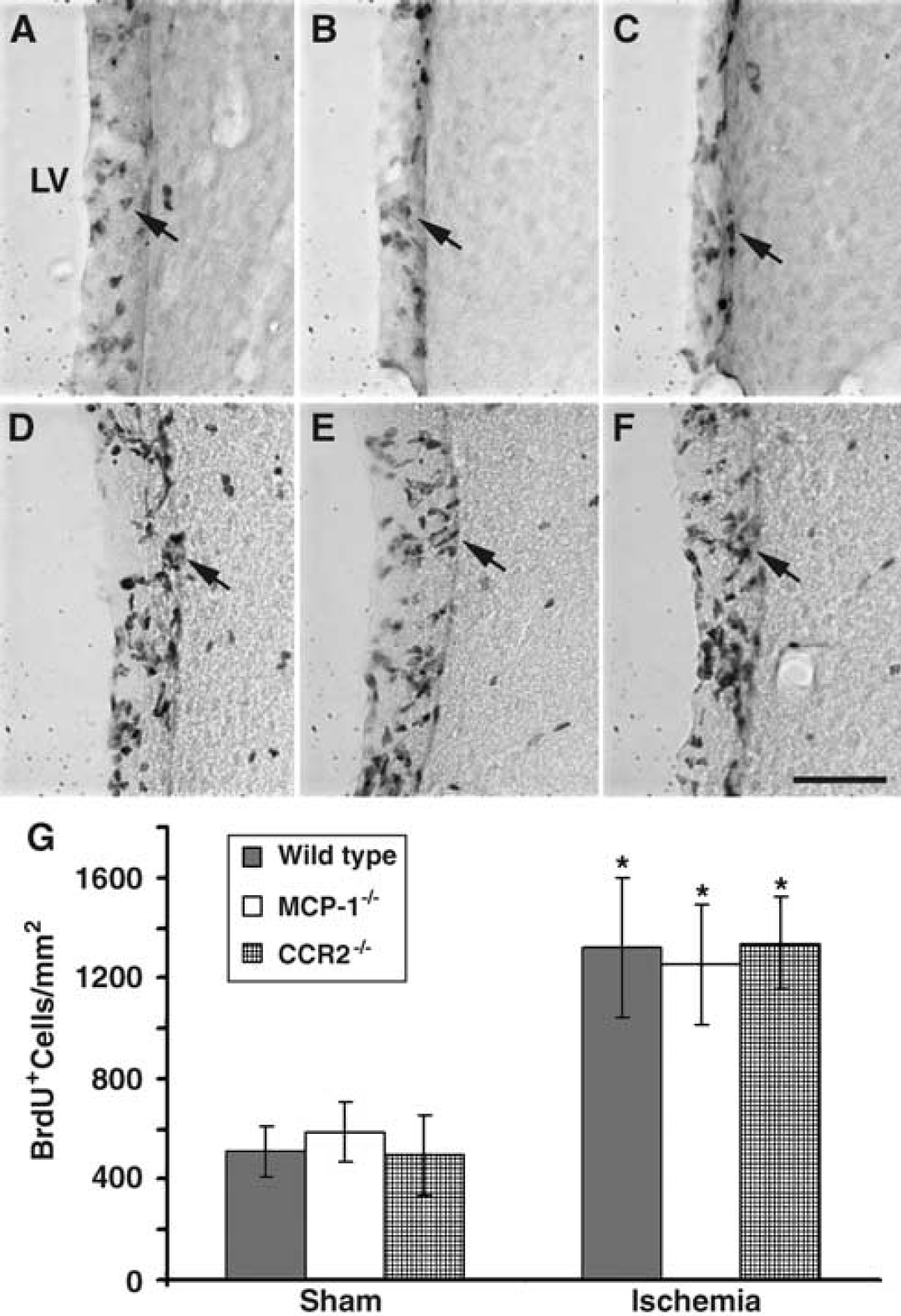
Deficiency in MCP-1 or CCR2 did not affect neural progenitor proliferation in the SVZ under normal and ischemic conditions. (
Discussion
Stroke is a major cause of disability among the adult population with relatively few therapeutic options once the damage has occurred. The discovery of neurogenesis in the adult mammalian brain has brought hope that this endogenous mechanism of cell replacement might be enhanced to repair the damaged brain. Although enhanced neurogenesis after cerebral ischemia was reported extensively (Dempsey et al, 2003; Zhang et al, 2005; Lichtenwalner and Parent, 2006), the mechanisms that mediate the various steps of neurogenesis (neural progenitor proliferation; neuroblast migration towards the injured area; differentiation, maturation and integration of newly generated neurons) are still not clear. We presently show that the chemokine MCP-1, produced by activated microglia and astrocytes in the ischemic area, attracts neuroblasts to migrate from the SVZ to the infarct.
Under normal conditions, the SVZ-derived neuroblasts migrate to the olfactory bulb via the rostral migratory stream. Although few newly formed neuroblasts migrate into the adjacent striatum from the SVZ in normal rodents, we observed that a large number of DCX positive neuroblasts migrated into the adjacent ischemic striatum after transient MCAO. We also observed that the SVZ-derived neuroblasts crossed the subcortical white matter and migrated into the ischemic cortex. In a global ischemic model, the pyramidal neurons in the CA1 region of the hippocampus are selectively damaged (Nakatomi et al, 2002). It has been shown that the PPV-derived neuroblasts migrate to the CA1 region and differentiate into mature pyramidal neurons after global ischemia (Nakatomi et al, 2002). Interestingly, we currently observed that the PPV-derived neuroblasts migrated in an opposite direction to the ischemic cortex after focal ischemia. Thus, the factors formed in the damaged tissue might attract neuroblasts to migrate towards it. This assumption is further supported by the observation that neural progenitors transplanted into the non-infarcted area of the ipsilateral cortex preferentially migrated towards the infarct.
Several previous studies have shown that the transplanted neural progenitors preferentially migrated to the damaged area of brain after focal ischemia (Zhang et al, 2003; Kelly et al, 2004; Jin et al, 2005), glioma invasion (Ehtesham et al, 2002) and autoimmune encephalomyelitis (Ben-Hur et al, 2003). Acute inflammation at the site of damage is the common feature of these disorders of various etiologies. In the injured brain, the activated microglia and astrocytes produce numerous factors such as growth factors, cytokines, and chemokines that are known to promote cell proliferation and migration. In this study, we observed that MCP-1 expression, which was localized in the activated microglia and astrocytes in the ischemic area, increased between 6 h to 3 days of reperfusion and returned to sham level by day 7 of reperfusion. MCP-1 is a chemokine of the CC family that binds to the G-protein-coupled receptor CCR2 on its target cells (Rossi and Zlotnik, 2000). It is a chemoattractant for monocytes/macrophages, T lymphocytes, basophils, and NK cells (Rossi and Zlotnik, 2000). Moreover, MCP-1 increases in vitro cultured neural progenitor migration across a chemotaxis chamber (Widera et al, 2004), with those progenitor cells expressing diverse chemokine receptors (Tran et al, 2004; Ji et al, 2004). Therefore, MCP-1 might be a potential factor responsible for the injury-initiated neural progenitor migration. We presently observed that when MCP-1 was infused into a normal rat striatum, the newly formed DCX positive neuroblasts, rather than after the rostral migratory stream, migrated from the SVZ towards the site of infusion. In addition, we also observed that neural progenitors from cultured neurospheres migrated towards the source of MCP-1 infused into the culture plate. The DCX positive cells in the SVZ and the migrating neuroblasts in the striatum expressed the MCP-1 receptor CCR2.
MCP-1 and CCR2 knockout mice subjected to transient MCAO showed significantly curtailed neuroblast migration from the SVZ to the ischemic striatum. This serves as a further evidence for the functional role of MCP-1 as one of the factors that cause progenitor migration in the post-ischemic brain. However, post-ischemic neuroblast migration was not completely blocked by the ablation of MCP-1 or CCR2. It is understandable that ischemia-induced neuroblast migration is a complicated process mediated by the synergetic action of many factors. Previous studies have shown that the chemokine stromal cell-derived factor (Imitola et al, 2004; Robin et al, 2006; Thoed et al, 2006), and matrix metalloproteinases (Lee et al, 2006; Wang et al, 2006) also play a role in the ischemia-induced neuroblast migration. A recent in vitro study also demonstrated that chemokines (especially MCP-1) attract the neural progenitors to migrate towards the site of neuroinflammation (Belmadani et al, 2006). Therefore, MCP-1 might be a chemoattractant for neural progenitor migration not only after ischemia but also in other neuroinflammatory diseases.
Previous studies showed exacerbation of ischemic brain damage in the MCP-1 transgenic mice and smaller infarcts in MCP1 knockout mice (Hughes et al, 2002; Chen et al, 2003). However, in the present studies neither MCP-1 knockout mice nor CCR2 knockout mice showed any significant difference in the infarct volume compared with wild-type controls mice. This discrepancy may be because the variations in the MCAO model (permanent versus transient) and the time of reperfusion at which the infarct volume was measured. The previous studies evaluated the infarct volume at 1 to 2 days after permanent MCAO. As the purpose of present study is to investigate ischemia-induced lateral neuroblast migration from the SVZ to the ischemic striatum, we used the transient MCAO model and measured the infarct volume at 5 days of reperfusion. The beneficial effect of MCP-1 deficiency on ischemic neural damage in the early time points after focal ischemia might be countered by other detrimental factors in the relatively later stages of ischemia.
In present study, we have demonstrated that MCP-1 promotes progenitor migration to help repair the damaged brain during a relatively later phase after focal ischemia. Thus, MCP-1 might have both beneficial and detrimental roles at different time points after focal ischemia. The signaling pathways by which MCP-1 mediates neural progenitor migration after ischemia need to be elucidated further. MCP-1 binds to its G-protein coupled receptor CCR2 on target cells to initiate diverse down-stream signaling events, such as induction of a pertussis toxin (PTX)-sensitive rise of intracellular calcium (Myers et al, 1995), inhibition of adenyl cyclase (Myers et al, 1995), stimulation of PI3-kinases (Turner et al, 1998), and activation of phospholipase C (Kuang et al, 1996) and extracellular signal-regulated kinases (Dubois et al, 1996). The PI3 kinase pathway has been shown to participate in the mediation of neuroblast migration after stroke (Katakowski et al, 2003). Elucidation of MCP-1 signaling pathways can result in developing reagents that can selectively induce neuroblast migration for brain repair after focal ischemia as well as other acute CNS insults.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.