
Abstract
This study explores the neuroprotective action of tumor necrosis factor-α (TNF-α) induced during physical exercise, which, consequently, reduces matrix metalloproteinase-9 (MMP-9) activity and ameliorates blood—brain barrier (BBB) dysfunction in association with extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation. Adult male Sprague—Dawley rats were subjected to exercise on a treadmill for 3 weeks. A 2-h middle cerebral artery occlusion and reperfusion was administered to exercised and nonexercised animals to induce stroke. Exercised ischemic rats were subjected to TNF-α inhibition and ERK1/2 by TNF-α antibody or UO126. Nissl staining of coronal sections revealed the infarct volume. Evans blue extravasation and water content evaluated BBB function. Western blot was performed to analyze protein expression of TNF-α, ERK1/2, phosphorylated ERK1/2, the basal laminar protein collagen IV, and MMP-9. The activity of MMP-9 was determined by gelatin zymography. Tumor necrosis factor-α expression and ERK1/2 phosphorylation were upregulated during exercise. Infarct volume, brain edema, and Evans blue extravasation all significantly decreased in exercised ischemic rats. Collagen IV production increased in exercised rats and remained high after stroke, whereas MMP-9 protein level and activity decreased. These results were negated and returned toward nonexercised values once TNF-α or ERK1/2 was blocked. We concluded that preischemic, exercise-induced TNF-α markedly decreases BBB dysfunction by using the ERK1/2 pathway.
Introduction
The blood-brain barrier (BBB) exists between the systemic circulatory system and the cerebral parenchyma and regulates substances entering the brain (del Zoppo et al, 1998). The extracellular matrix (ECM) of the BBB forms a basal lamina, which surrounds and anchors endothelial cells and astrocytes, which, in turn, provide a structural barrier that selectively filters blood elements. Major components of the cerebral microvascular basal lamina include collagen IV, laminin, and fibronectin (Yurchenco and Schittny, 1990). After middle cerebral artery occlusion, the primary vascular permeability barrier is lost and alterations occur in the cerebral microvascular basal lamina (del Zoppo and Mabuchi, 2003). When the BBB is compromised, vascular exudates leak into the surrounding tissues causing detrimental swelling in the cranial cavity.
An earlier study showed that ischemic rats with physical exercise had a reduction in infarct, which correlated with reduced brain edema (Wang et al, 2001). In addition, previously we reported that physical exercise improved microvascular integrity and decreased permeability of the BBB after a stroke (Ding et al, 2006). However, we have yet to delineate the mechanisms by which exercise improves damaged BBB function after stroke-induced injury.
Proteins and polysaccharides that make up the ECM are degraded by a variety of extracellular proteolytic enzymes, which includes the matrix metalloproteinase (MMP) family. Matrix metalloproteinases, which are produced by endothelial cells, microglia, and astrocytes, are divided into five classes based on in vitro substrate analysis, including gelatinase B (MMP-9) (Lo et al, 2003). Increasing evidence indicates that there is an upregulation of MMP-9 after the onset of focal ischemia and reperfusion (I/R), which is associated with an alteration of BBB permeability and the formation of vasogenic edema.
Tumor necrosis factor-α (TNF-α) has been determined to be a deleterious cytokine in stroke and contributes to brain inflammation and neuronal damage (Hallenbeck, 2002). Conversely, increasing evidence suggests the salutary roles of TNF-α in tissue repair and in the evolutionary mechanism fundamental for survival and neuroprotection (Cheng et al, 1994; Bruce et al, 1996; Nawashiro et al, 1997). Still, additional evidence suggests that TNF-α acts as a trigger in classic ischemic preconditioning, evidenced by a significant induction of TNF-α mRNA expression in brains subjected to an ischemic preconditioning procedure leading to neural tolerance (Bruce et al, 1996; Nawashiro et al, 1997; Ginis et al, 1999; Liu et al, 2000). Our recent report (Ding et al, 2005) showed that mRNA expression of TNF-α increased after exercise and reached significant levels at 2 and 3 weeks. However, little is known about the mechanisms by which TNF-α prevents ischemic injury, particularly in BBB dysfunction, and its transduction signaling pathway.
Extracellular signal-regulated kinase-1 and −2 (ERK1/2) (Sharony et al, 2006), the most extensively characterized mitogen-activated protein kinase pathways, play a pivotal role in signal transduction during neuropathologic processes responding to I/R injury. Growing evidence implicates that ERK signaling plays a beneficial and protective role in many systems, and that activation of ERK1/2 is an important defense mechanism against transient hypoxia/ischemia because of its ability to counteract cell death and enable damage repair (Hetman and Gozdz, 2004). Currently, a few studies identified the interaction between phosphor-ERK1/2 and TNF-α (Lee et al, 2001, 2005; Gortz et al, 2005). In this study, we determined whether preischemic exercise exhibits its neuroprotective function through activation of mitogen-activated protein kinase kinase/ERK signaling pathways, and whether preischemic expressions of phosphor-ERK1/2 and TNF-α are associated.
Materials and methods
Subjects
Sprague—Dawley rats (260 to 300 g, 3-month-old, Charles River, Wilmington, MA, USA) were housed in the same animal care facility during a 12-h light/dark cycle throughout the study. Animal care and surgical procedures were performed in accordance with the guidelines approved by the NIH and the University of Texas Health Science Center at San Antonio Animal Investigation Committee.
Motor Exercise
Animals were randomly assigned to treadmill exercise or nonexercise control groups. All animals ran on a four-lane treadmill (AccuPacer, AccuScan Instruments Inc., Columbus, OH, USA) at a speed of 30 m/min for 30 mins each day, 5 days/week. Animals that were not willing to run were excluded from further study (n = 4).
Inhibition of TNF-α and ERK1/2 Activity
A polyclonal rabbit anti-mouse TNF-α neutralizing antibody (0.30 mg/kg; Genzyme, Cambridge, MA, USA) dissolved in nonpyrogenic sterile saline was injected intravenously into rats in the exercise group at 24 and 48 h before I/R. 1,4-Diamino-2,3-dicyano-1,4-bis-[2-aminophenylthio]-butadiene (U0126) is a potent inhibitor of the dual specificity of mitogen-activated protein kinase kinase 1 and 2, the activator and upstream kinase of ERK1/2. A dose of 0.2 mg/kg of U0126 (Calbiochem, La Jolla, CA, USA) dissolved in 1% dimethyl sulfoxide was administered intravenously to the exercise group at 10 mins before middle cerebral artery occlusion and 10 mins before reperfusion.
Induction of Stroke with an Intraluminal Filament
Stroke was induced in 3-week exercised and nonexercised rats. Animals were anesthetized and maintained with 1% to 3% isoflurane in 70% N2O and 30% O2 with a facemask. Rectal temperature was maintained at 37°C with a circulating heating pad. Middle cerebral artery occlusion was induced using an intraluminal filament model. The reliability and effectiveness of this model to induce stroke was guaranteed by using poly-
Physiologic variables, such as blood pressure, blood gases (pH, pO2, pCO2), and hematocrit, were monitored before and after surgical procedures through the cannulated right femoral artery.
Infarct Volume
Ischemic brains, 48 h after reperfusion, were sectioned coronally at a thickness of 10 μm from 2.0 to −4.0 mm bregma and were processed for Nissl staining to evaluate infarct volume in exercised rats, TNF-α neutralizing antibody-treated exercised rats, ERK inhibitor-treated exercised rats, and nonexercised rats (n = 5 × 4).
Evaluation of BBB Disruption
Brain edema was evaluated by measuring water content. Brain tissue samples from exercised ischemic rats with or without ERK inhibitor or TNF-α treatment and from nonexercised ischemic rats (n = 5 × 4) were immediately weighed to obtain wet weight (WW). The tissue was then dried in an oven at 70°C for 72 h and weighed again to obtain the dry weight (DW). The formula (WW—DW)/WW × 100% was used to calculate the water content and expressed as a percentage of wet weight.
Blood—brain barrier permeability was determined by measuring the amount of Evans blue extravasation from the ipsilateral and contralateral hemispheres. Forty-six hours after reperfusion, Evans blue dye (2%, 4 mL/kg body weight) was slowly administered intravenously and allowed to circulate for 2 h in exercised rats with or without ERK inhibitor or TNF-α antibody treatment, as well as nonexercised ischemic rats (n = 5 × 4). At the end of the experiment, the rats were perfused with saline to wash away any remaining dye in the blood vessels, and the two hemispheres of each brain were dissected and frozen at −80°C for analysis. The Evans blue dye was extracted by homogenizing the sample in 3.5 mL of 0.1 mol/L phosphate-buffered saline at pH 7.4. To precipitate the protein, 2.5 mL of 60% trichloroacetic acid was added. The mixture was vortexed for 2 mins and cooled for 30 mins. To pellet the brain tissue, the sample was centrifuged for 40 mins at 1,000 r.p.m. The absorption of the supernatant was measured at 610 nm with a spectrophotometer (AD 340, Beckman Coulter, Fullerton, CA, USA). The content of Evans blue from the two hemispheres was expressed as μg/mL of brain tissue by using a standardized curve.
Western Blot Evaluation of TNF-α, ERK1/2, Phosphorylated ERK1/2, Collagen IV, and MMP-9
Western blot analysis was used to quantitatively detect TNF-α protein expression in exercised and control animals (n = 5 × 2), collagen IV and MMP-9 protein expression in ischemic exercised rats with or without ERK inhibitor or TNF-α antibody treatment, and in nonexercised ischemic rats (n = 5 × 4). Equal amounts of protein (30 μg/well) were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked for 1 h at room temperature with 5% skim milk in TBST and then incubated with the primary antibodies (rabbit polyclonal anti-TNF-α antibody, dilution 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA; rabbit polyclonal anti-ERK1/2 antibody, dilution 1:1,500, Cell Signaling, Danvers, MA, USA; rabbit polyclonal anti-phosphor-ERK1/2 antibody, dilution 1:2,000, Cell Signaling; rabbit polyclonal anti-collagen IV antibody, dilution 1:400, Santa Cruz Biotechnology; and mouse monoclonal anti-MMP-9 antibody, dilution 1:2,000, Sigma, St Louis, MO, USA) overnight at 4°C. After incubation with the secondary antibodies for 1 h at room temperature, detection of immunoreactive bands was performed with the ECL system (GE Healthcare, Piscataway, UK). The supernatants were used as whole-tissue lysates and protein concentration was determined using the Bradford assay (Bio-Rad). Protein equal loading was confirmed by intracellular protein β-actin (goat polyclonal anti-β-actin antibody, dilution 1:1,000, Santa Cruz Biotechnology). The intensity of protein expression was quantified using the Chemi-GeniusQ Imaging Analysis System.
Evaluation of TNF-α Protein Expression by Enzyme-Linked Immunoabsorbent Assay
Enzyme-linked immunoabsorbent assay was further performed for detecting cerebral levels of TNF-α protein expression (n = 5 × 2), using a commercially available rat TNF-α/TNFSFIA ELISA kit (R&D Systems, Minneapolis, MN, USA). Dilution curve of the protein samples was parallel to the standard dilution curve. The assays were performed according to the manufacturer's recommended procedures. The absorbance at 450 nm was measured with an automatic wavelength correction of 570 nm.
Gelatin Zymography of MMP-9 Activity
To determine the enzymatic activity of MMP-9, gelatin zymography was performed, as previously described by us (Swann et al, 2007), in exercised ischemic rats with or without ERK inhibitor or TNF-α antibody treatment and in nonexercised ischemic rats (n = 5 × 4). The samples (10 μL per well) were applied on Novex zymogram gels (Invitrogen, Carlsbad, CA, USA) and the gels were electrophoresed, developed, and stained.
Statistical Analysis
All the data were described as mean ± s.e. Statistical analysis was performed with SPSS for Windows, version 15.0 (SPSS Inc.). The differences between two groups were assessed using a two-tailed independent t-test with a significance level at P < 0.05. The differences among multiple groups were assessed using one-way analysis of variance with a significance level at P < 0.05. Post hoc comparison between groups was further detected using the least significant difference method.
Results
Arterial blood pressure, blood pH, hematocrit, and blood gases of the animals remained comparable among the groups (data not shown).
Protein Expressions of TNF-α and Phosphorylated ERK1/2
Western blotting showed a significant (P < 0.01) increase in TNF-α levels (3.01 ± 0.24) in exercised animals for 3 weeks compared with animals in the control group, which were assigned a value of 1.0 ± 0.0 (Figure 1). A sensitive enzyme-linked immunoabsorbent assay analysis further showed protein expression of TNF-α in brain tissue from rats with or without exercise (Figure 2). The result showed that exercised rats display significantly (P < 0.01) higher (11-fold) concentrations (43.9 ± 4.4 pg/mg total protein) of TNF-α in comparison with very low concentrations (4.1 ± 0.8 pg/mg total protein) in the nonexercise control group. At the same time point, ERK1/2 activity levels in the brain sample from exercised animals were determined by western blot with a phosphor-ERK1/2 antibody, showing a significant (P < 0.01) increase in ERK1/2 phosphorylation levels by 65 ± 18% (Figure 3A) but not in total ERK1/2 level (Figure 3B). This suggests a temporal association of TNF-α expression and ERK1/2 activation.
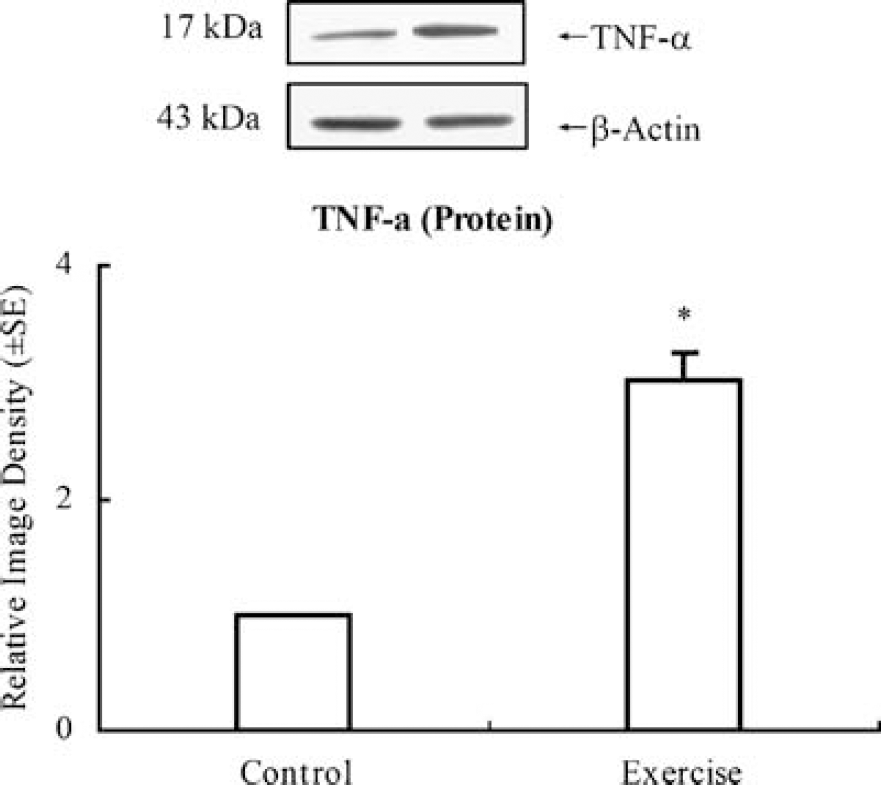
Tumor necrosis factor-α western blot and graph depicting a significant increase after 3-week exercise over control (P < 0.05, indicated by *). Representative immunoblots are presented. Western blot of β-actin showed equal loading of protein in each lane of the gel.
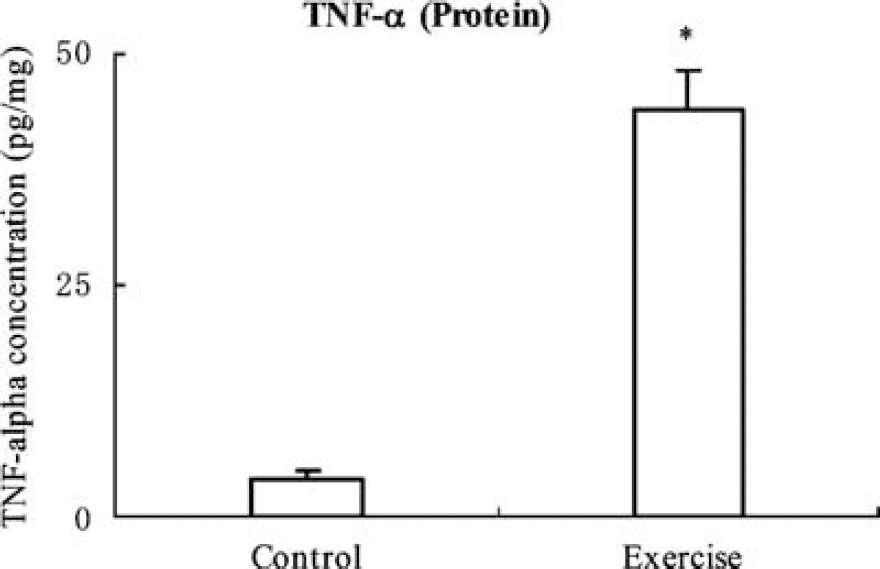
Upregulation of TNF-α protein levels was shown by enzyme-linked immunoabsorbent assay in a rat exercised for 3 weeks. The analysis shows a significantly (P < 0.01, indicated by *) higher (11-fold) concentration (43.9 ± 4.4 pg/mg total protein) of TNF-α in exercised rats compared with very low concentrations (4.1 ± 0.8 pg/mg total protein) in the nonexercise controls.
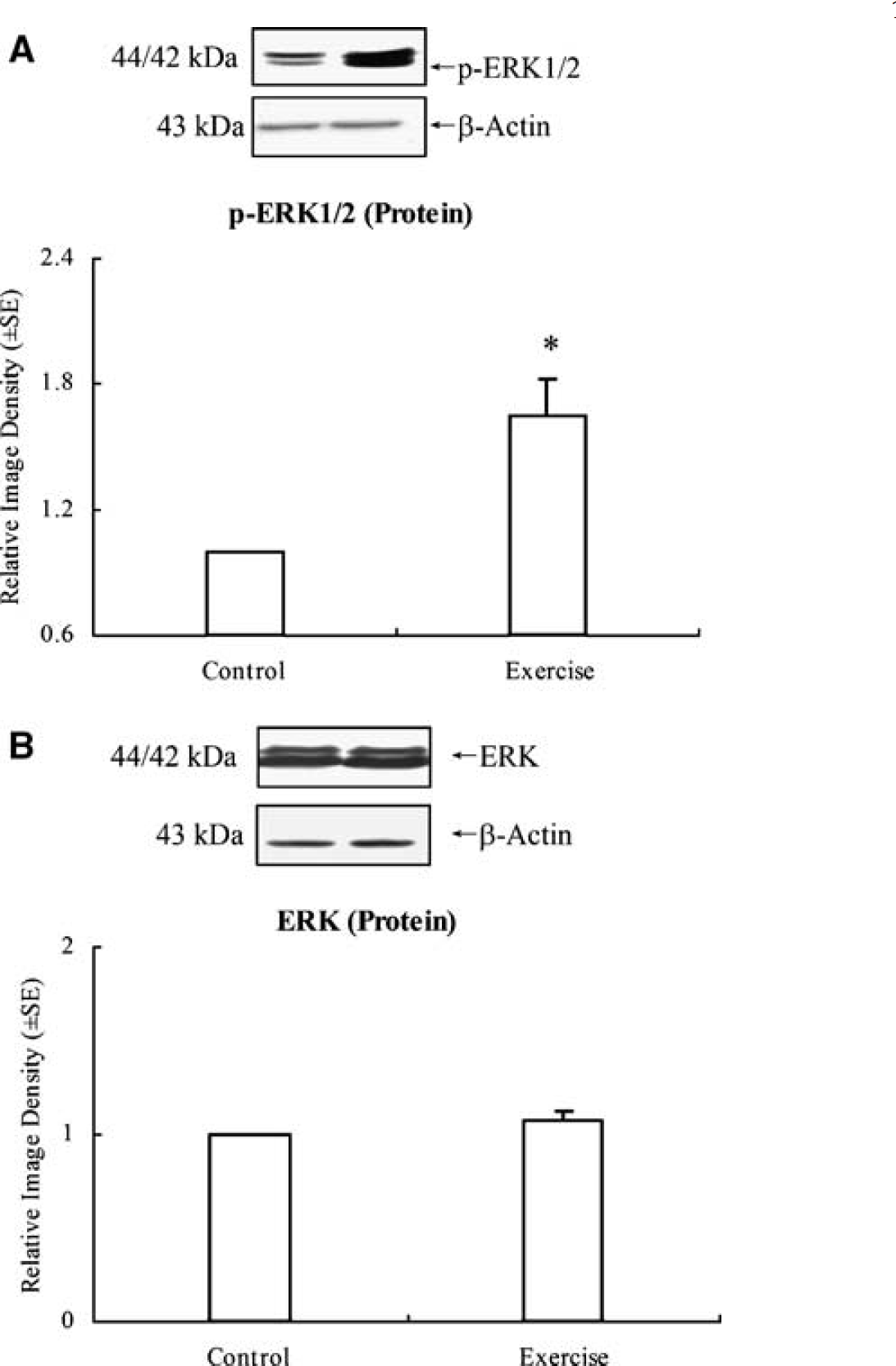
After 3-week exercise, ERK1/2 activity levels were determined by western blot with a phosphor-ERK1/2 antibody. The result shows a significant (P < 0.01) increase in ERK1/2 phosphorylation levels (by 165 ± 18%) (
Infarct Volume
The infarct region, defined as the area with reduced Nissl staining or containing dark, pyknotic-necrotic cell bodies (Figure 4A), was determined by subtracting the noninfarcted region in the ipsilateral hemisphere from that in the contralateral hemisphere. The infarct volume was thus presented as a percentage of the volume of the contralateral hemisphere (Figure 4B). Infarct volume was significantly (F(3,16) = 74.1, P < 0.01) decreased by 82% in exercised ischemic animals (8.8 ± 1.7%) compared to nonexercise ischemic animals (49.5% ± 2.3%). However, when TNF-α or ERK1/2 activity was inhibited, infarct volumes were reversed to higher levels (43.49% ± 3.12% or 50.49% ± 5.16%) and neutralized 80% and 83% of exercise-induced protection, respectively (P < 0.01).
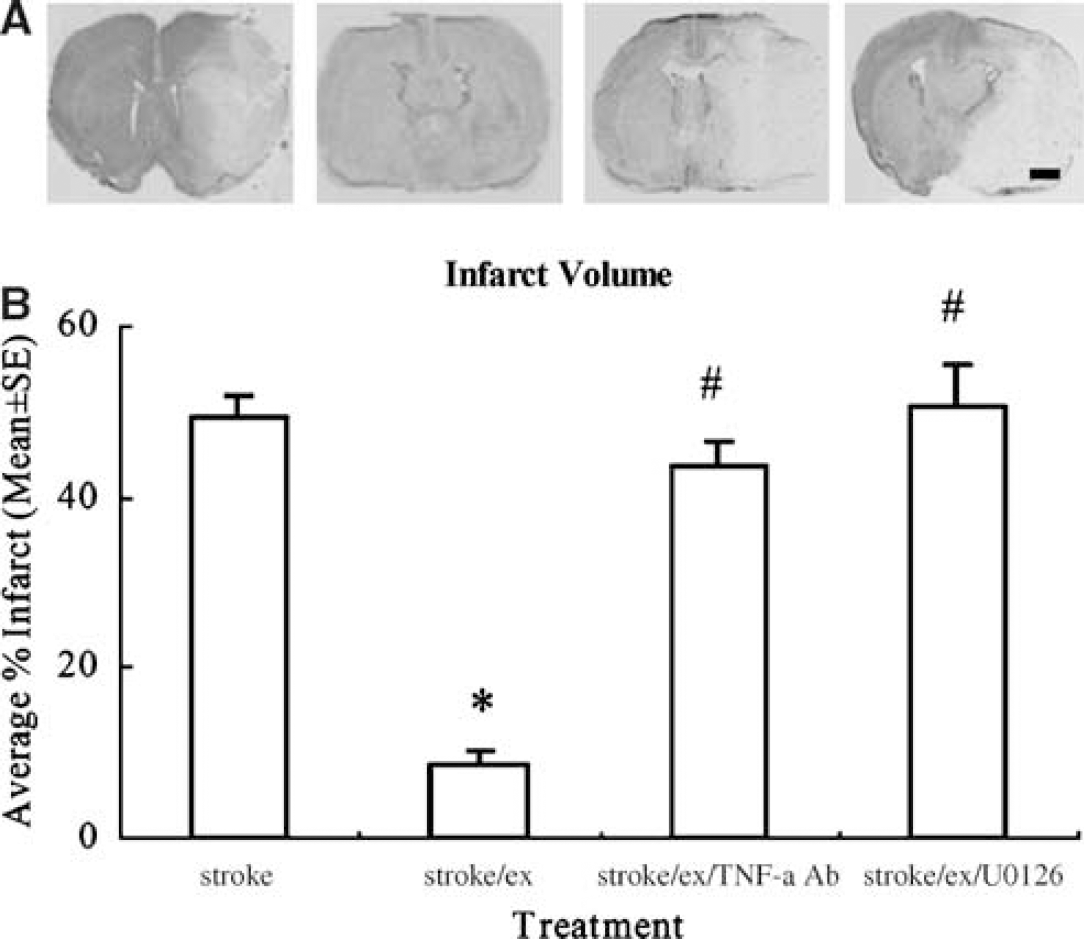
(
BBB Permeability and Integrity
Brain edema, determined by water content, was significantly (F(3,16) = 6.3, P < 0.01) less in exercised ischemic rats compared to that in ischemic rats without exercise. As compared to 79% water content in control subjects, nonexercised ischemic subjects had 85.6 ± 1.2% water content in brain tissue, whereas exercised ischemic subjects had only 80.9 ± 0.4% water content. The results indicate a 71% reduction in brain edema in I/R injured rats after physical exercise. However, in exercised ischemic animals in which TNF-α or ERK1/2 activities were inhibited by polyclonal rabbit anti-mouse TNF-α neutralizing antibody or U0126, the reduction in the level of brain edema as a result of exercise was significantly (P < 0.05) reversed (Figure 5), with 85.1 ± 0.5% and 85.77 ± 1.82% water content.
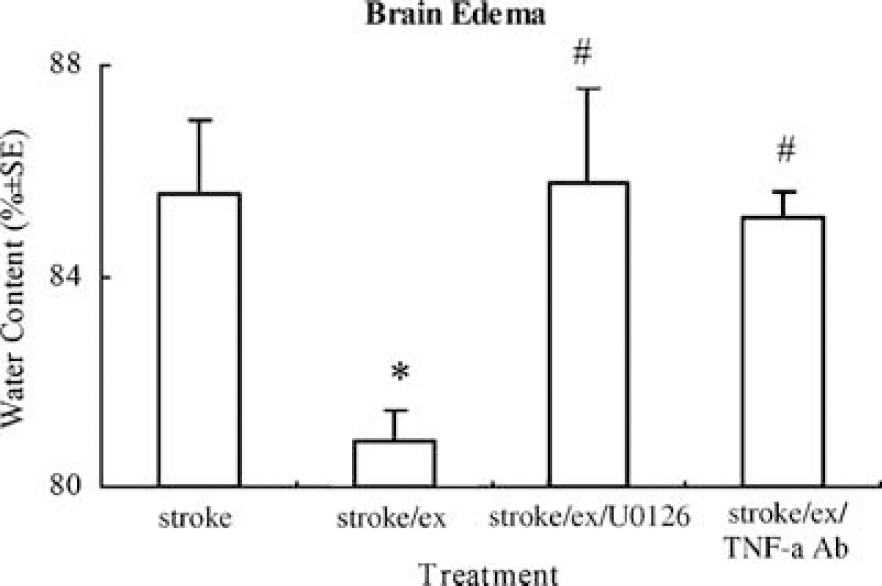
Brain edema after stroke determined by water content significantly (P < 0.05, indicated by *) decreases in exercised versus nonexercised rats. Blocking TNF-α or ERK1/2 significantly (P < 0.05, indicated by #) annuls the reduction in brain edema, with water content returning to the levels in nonexercised ischemic rat.
Blood—brain barrier permeability was determined by Evans blue dye extravasation. Increased BBB permeability after stroke in the ipsilateral hemisphere as compared with the contralateral hemisphere (24.3 ± 6.1 versus 4.6 ± 0.3 μg/mL) was significantly (P < 0.01) reduced by preischemic exercise as shown by the small amount of Evans blue concentration (4.57 ± 0.38 versus 3.65 ± 0.28 μg/mL) in the brain tissue (Figure 6). Specifically, nonexercised ischemic rats had a considerable loss of BBB integrity and an increase in BBB permeability compared to exercised ischemic rats as shown by the levels of Evans blue (24.3 ± 6.1 versus 4.57 ± 0.38 μg/mL). There was a significant (F(3,16) = 10.1, P < 0.01) reversal in Evans blue content in rats in which TNF-α and ERK were inhibited after 2-h middle cerebral artery occlusion and 48 h reperfusion in the ipsilateral hemisphere as compared with the contralateral hemisphere (17.1 ± 4.0 versus 6.4 ± 0.4 μg/mL and 13.8 ± 0.5 versus 7.0 ± 0.6 μg/mL, respectively).
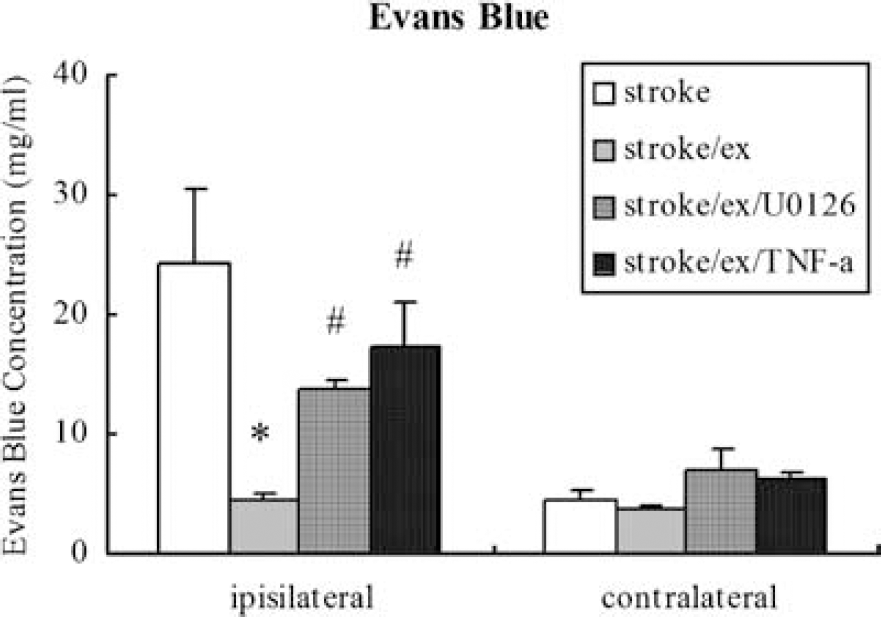
Similar to the results of infarct volume, there is a significant (P < 0.05, indicated by *) reduction in Evans blue extravasation in the ipsilateral hemisphere of exercised ischemic rats compared to that in nonexercise controls. The reduced levels of Evans blue leakage after preischemic exercise significantly (P < 0.05, indicated by #) return toward that of nonexercised ischemic rat after blocking ERK1/2 and TNF-α. The overall effect on the contralateral side is minimal among groups.
Expressions of Collagen IV and MMP-9
Collagen- IV protein levels for the control group were arbitrarily assigned 1.0 ± 0.0 to serve as reference. Collagen IV relative protein levels decreased significantly (P < 0.01) after I/R injury in nonexercised subjects (Figure 7). Preischemic exercise significantly (P < 0.01) enhanced the expression of collagen IV protein. Inhibition of TNF-α activity by its neutralizing antibody or ERK1/2 activity by U0126 significantly (F(3,16) = 4.4, P < 0.05) decreased collagen IV expression toward nonexercised levels.
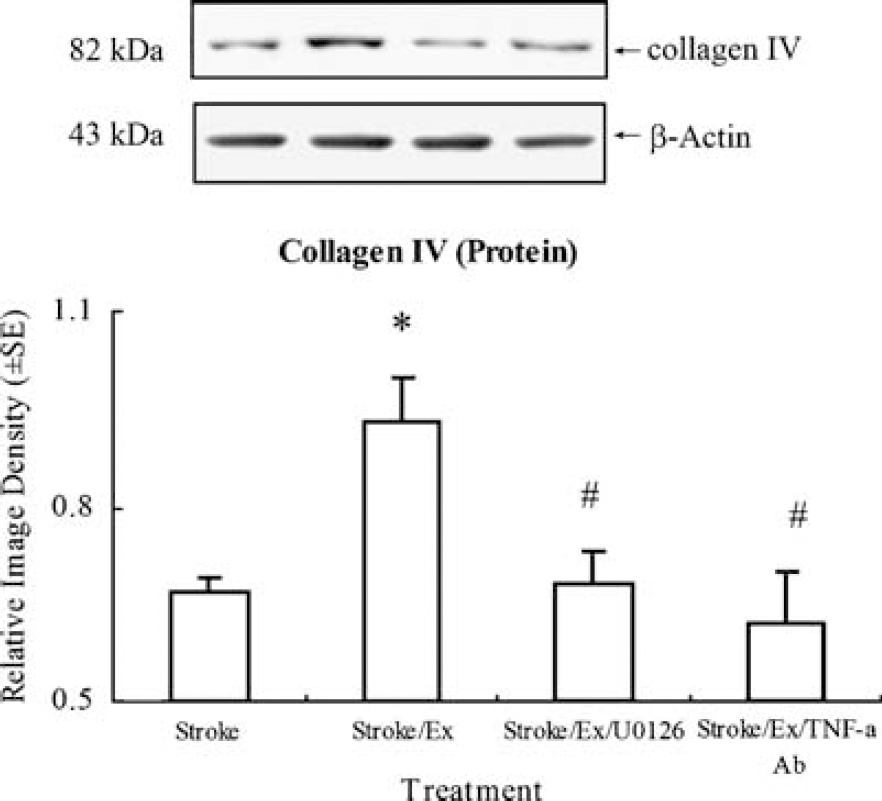
As compared to control levels arbitrarily assigned 1.0 ± 0.0 (not shown) to serve as reference, exercised ischemic rats show a significant (P < 0.05, indicated by *) increase in collagen IV expression over nonexercised ischemic rats. Inhibiting TNF-α or ERK1/2 with TNF-α antibody or UO126 significantly (P < 0.05, indicated by #) reverses the increase in collagen IV seen in the exercised rat to the level in ischemic rats without preischemic exercise. Representative immunoblots are presented.
The expression and activity of MMP-9 were measured to determine the effects of TNF-α upregulation and ERK1/2 activation on MMP-9-associated brain damage. As compared to the control group, the stroke group showed an increased MMP-9 protein expression. Also, preischemic exercise significantly reduced MMP-9 expression (F(3,16) = 16.0, P < 0.01). This study further indicated that inhibition of TNF-α or ERK1/2 in exercised, ischemic animals significantly (P < 0.01) increased MMP-9 protein expression levels (Figure 8).
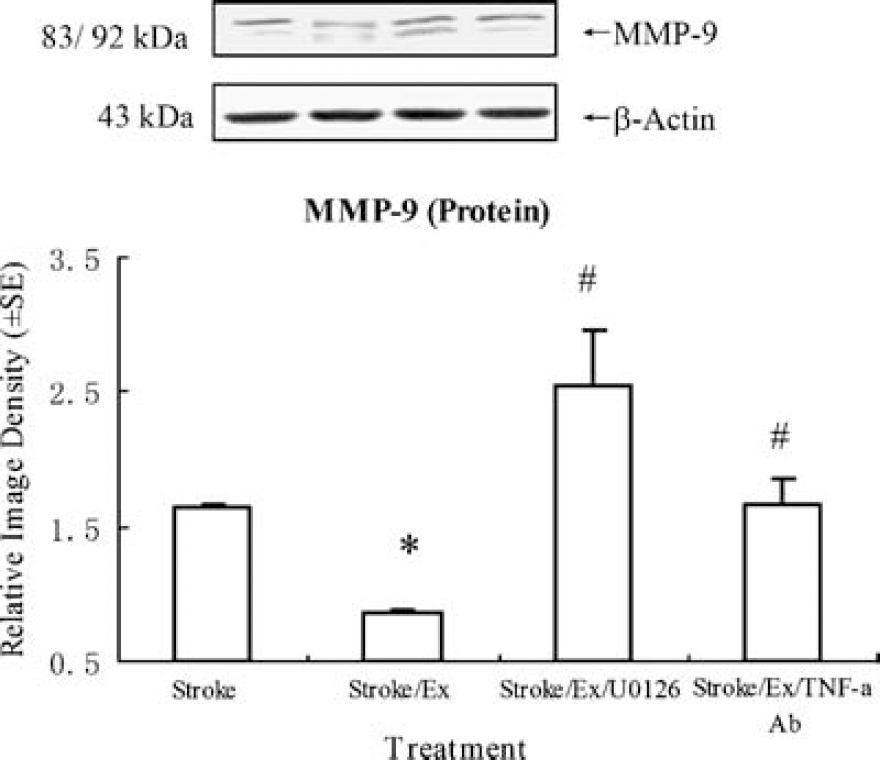
Preischemic exercise significantly (P < 0.05, indicated by *) reduces the increase in MMP-9 protein during stroke. This effect is inhibited significantly (P < 0.05, indicated by #) by TNF-α antibody or UO126. Furthermore, an additional increase in MMP-9 protein levels is seen in rats in which ERK1/2 is inhibited. Representative immunoblots are presented.
Gelatin zymography showed a significant (F(3,16) = 26.8, P < 0.01) increase in MMP-9 activity shown by a relative image density of 5.7 ± 0.1 in nonexercised, ischemic rats versus 0.5 ± 0.1 in exercised, ischemic rats (Figure 9). Control levels were arbitrarily assigned 1.0 ± 0.0 to serve as a reference. Relative MMP-9 enzyme activity was significantly increased in exercised, ischemic rats with TNF-α inhibition, as shown by image density values of 2.7 ± 0.6. Furthermore, after the administration of the ERK1/2 inhibitor, the reduction in MMP-9 activity previously seen in preischemic exercise was significantly (P < 0.01) reversed, as shown by image density values of 2.85 ± 0.22.
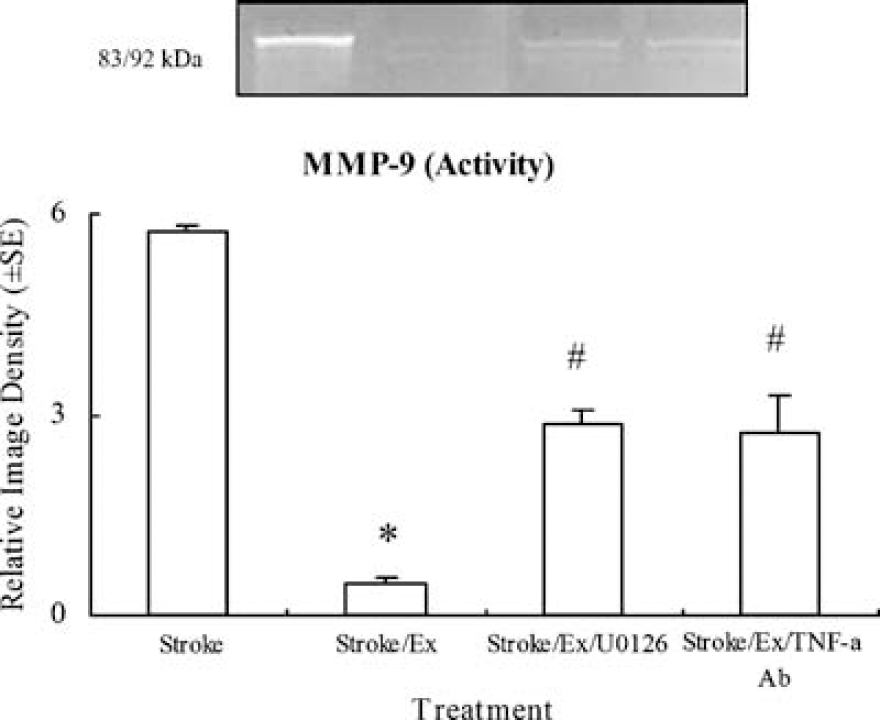
Zymograph and graph showing that preischemic exercise significantly (P < 0.05, indicated by *) reduces the increase in MMP-9 enzyme activity caused by stroke. Inhibition of TNF-α or ERK1/2 with TNF-α antibody or UO126 significantly (P < 0.05, indicated by #) reverses the decreased levels of MMP-9 activity by exercise in exercised ischemic rats.
Discussion
This study shows that physical exercise effectively reduces brain infarct volume after focal ischemia, which is associated with improved BBB integrity. Preischemic induction of TNF-α by physical exercise, as well as ERK1/2 signaling, plays a key role in reducing BBB dysfunction in stroke. This enhanced BBB function correlated with an enhanced basal lamina indicated by collagen IV expression in ischemic rats with exercise. The expression of MMP-9 after stroke was reduced by preischemic exercise, which would play a beneficial role in collagen IV expression against I/R injury. Our study illustrates that these favorable outcomes, induced during exercise, are eliminated by the inhibition of either TNF-α or ERK1/2 activation.
TNF-α in Exercise-Reduced Ischemic Injury
Tumor necrosis factor-α has been determined to be a deleterious cytokine in stroke. Cerebral ischemia causes an acute upregulation of TNF-α and TNF receptors in neurons, astroglia, and leukocytes, as well as in endothelial cells, leading to brain inflammation and neuronal damage (Hallenbeck, 2002). Conversely, there has been an increasing amount of evidence suggesting the salutary roles of TNF-α in tissue repair and as an evolutionary mechanism fundamental for survival and neuroprotection (Cheng et al, 1994; Bruce et al, 1996; Nawashiro et al, 1997). Previous studies have showed that pretreatment with TNF-α reduces the inflammatory reaction (intercellular adhension molecule-1 expression) in cortical astrocyte cultures after ischemic stress (Ginis et al, 2002). Additional evidence suggests that TNF-α acts as a trigger of classic ischemic preconditioning, with a significant induction of TNF-α mRNA expression in brains subjected to an ischemic preconditioning procedure leading to neural tolerance (Bruce et al, 1996; Nawashiro et al, 1997; Ginis et al, 1999; Liu et al, 2000). Pharmacologic pretreatment of TNF-α in stroke subjects effectively reduced brain infarction in rodent models (Nawashiro et al, 1997). Our recent study indicates that during the course of exercise preconditioning, expression of TNF-α mRNA progressively increased after 2 and 3 weeks of exercise (Ding et al, 2005). This study further showed that physical exercise significantly increased protein expression of TNF-α. This study also showed a pivotal role of exercise-induced upregulated TNF-α in reducing infarct volume, BBB disruption, loss of basal laminar protein, and MMP expression after stroke by eliminating neuroprotection with inhibition of TNF-α. Together, these studies suggest that TNF-α, if generated before ischemic insult, could have a beneficial contribution in stroke. This powerful neuroprotection of exercise-induced TNF-α may involve intracellular location, concentration, and its interactions with other molecules elicited during exercise. The mechanisms by which TNF-α expression prevents BBB disruption may not only enhance our understanding of mechanisms underlying exercise-induced neuroprotection, but also provide a better insight into the therapeutic potential of TNF-α as a candidate for pharmacologic intervention.
Role of ERK1/2 in Ischemic Injury
The ERK1/2 pathway plays a pivotal role in signal transduction in neuropathologic processes responding to I/R injury. Previous work has shown a death-promoting role of ERK in neurons and other cell types (Chu et al, 2004; Zhuang and Schnellmann, 2006). On the other hand, growing evidence suggests that ERK signaling plays a beneficial, protective role in many systems (Hetman and Gozdz, 2004; Sawatzky et al, 2006). Activation of ERK1/2 is an important defense mechanism against transient hypoxia/ischemia that attempts to counteract cell death and damage repair. Induction of ischemic tolerance after ischemic preconditioning is modeled by activation of ERK1/2 in cultured rat cortical neurons (Gonzalez-Zulueta et al, 2000), as well as in the in vivo ischemic brain (Shamloo and Wieloch, 1999; Jones and Bergeron, 2004), suggesting that phosphorylation of ERK1/2 before fatal stroke has neuroprotective roles. Phosphorylation of ERK1/2 is also increased in brain areas that survive ischemia, such as in the penumbra after focal ischemia in rats (Kitagawa et al, 1999) and in the ischemia-resistant dentate gyrus and CA3 regions of the hippocampus after global ischemia (Hu and Wieloch, 1994; Hu et al, 2000). Survival signaling cascades may become activated and initiate neuroprotective processes (Hetman and Gozdz, 2004), whereas ERK1/2 signaling has a detrimental role in cerebral ischemia, brain trauma, and neurodegenerative diseases (Chu et al, 2004). In a recent study using a rat heart I/R injury model, ischemic preconditioning activated ERK1/2 (Lecour et al, 2005). In this study, reversal of exercise-induced neuroprotection by inhibiting ERK1/2 activity suggested a beneficial role of ERK1/2 in exercise-reduced ischemic injury, which was associated with preischemic expression of TNF-α. Under exercise preconditioning, phosphor-ERK1/2 could contribute beneficially to stroke. Currently, only very few studies have examined the interaction between phosphor-ERK1/2 and TNF-α (Lee et al, 2001, 2005; Gortz et al, 2005). Tumor necrosis factor-α was reported to promote the survival of osteoclasts by involving ERK1/2 activity. The limitation of this study included lack of understanding of whether TNF-α is involved in ERK1/2 phosphorylation or whether the protective effects of TNF-α are mediated by ERKs. A cross-talk between TNF-α and signaling ERK1/2 activation needs further investigation.
Detrimental Roles of MMP-9 in BBB Disruption After Cerebral I/R Injury
Clinically, the disorders after ischemic insults lead to disruption of the BBB with concomitant vasogenic edema and hemorrhagic transformation, as well as disturbance of homeostasis within the brain microenvironment (Wang and Lo, 2003). Brain microvessel integrity requires maintenance of the endothelial permeability barrier and basal lamina derived from ECM. The ECM not only provides structural support for cells, but also acts as a physical barrier to or as a selective filter for soluble molecules. Focal ischemia abruptly alters the stable relationships among endothelial cells, astrocytes, and the intervening ECM. The endothelial cells—astrocyte—matrix interactions provide the central trigger for initiation of brain injury during a stroke (Petty and Wettstein, 2001). Therefore, enhanced ECM as a result of upregulation of collagen IV after physical exercise in this study may play a key role in reducing brain edema and BBB permeability. Inhibition of either TNF-α or ERK1/2 reverses the reduced BBB dysfunction and collagen IV expression, suggesting that the TNF-α-ERK1/2 cascade plays a beneficial role in exercise-induced neuroprotection.
Previous evidence has indicated that MMP-9 produced in endothelial cells, microglia, and astrocytes is upregulated after the onset of permanent (Romanic et al, 1998; Gasche et al, 1999) or transient (Rosenberg et al, 1998; Heo et al, 1999) focal ischemia in experimental animals, as well as in human patients (Clark et al, 1997; Horstmann et al, 2003). In the central nervous system, the early appearance of activated MMP-9 has been associated with alterations in BBB permeability or degradation of critical BBB components and the formation of vasogenic edema after transient focal ischemia (Rosenberg et al, 1998; Heo et al, 1999). In addition, pharmacologic inhibition of MMPs ameliorated the edema after focal cerebral ischemia (Romanic et al, 1998; Rosenberg et al, 1998), and MMP-9-deficient knockout mice showed reduced BBB disruption and edema after transient focal cerebral ischemia (Asahi et al, 2001) and traumatic brain injury (Wang et al, 2000). These results support the notion that a reduction in MMP expression induced by preischemic exercise after stroke is attributable to improved BBB function. In this study, BBB disruption after stroke lessened by preischemic exercise correlates with levels of MMP-9 expression (protein and enzyme activity). The reduction in BBB disruption by physical exercise was largely reversed by inhibiting TNF-α and ERK1/2 activation, suggesting the role of the TNF-α–ERK1/2 cascade in exercise-induced neuroprotection.
A few studies have documented a correlation between cytokines and MMP-9 expression. An associated increase in mRNA levels of MMP-9 and TNF-α, as well as an increase in their protein synthesis, was reported in delayed-type hypersensitivity lesions in the brain (Anthony et al, 1998). Previous studies indicate that acute increases in TNF-α cause MMP-9 expression through ERK activation (Arai et al, 2003; Hosomi et al, 2005). In contrast, the present data might suggest that preischemic TNF-α upregulation, in association with physical exercise, significantly reduced MMP-9 expression through ERK1/2 signaling. Although TNF-α and ERK1/2 inhibition reversed the reduction in MMP-9 expression by preischemic exercise, direct evidence showing the role of the TNF-α-ERK1/2 cascade in reducing MMP-9 needs to be further investigated, such as whether ERK1/2 activation is altered by the moderate increases in TNF-α during exercise, leading to a reduction in MMP-9 activity in response to I/R injury.
This study shows a correlative relationship between physical exercise and reduced disruption of the BBB, which was determined by Evans blue extravasation, brain edema percentage, and collagen IV expression. This study also explains that exercise-induced neuroprotection is attributable to TNF-α expression and ERK1/2 activation. This finding would not only clarify mechanisms of exercise-induced neuroprotection, but also help develop new strategies for stroke therapy. An in-depth understanding of the role of the TNF-ERK cascade in regulating BBB dysfunction and neuronal damage after stroke may offer better understanding of the molecular constituents in exercise-reduced I/R injury and uncover novel, attractive, and potent targets for stroke therapy.
Footnotes
Acknowledgements
We are grateful to Mr Yandong Zhou for his help in preparation of the manuscript. This work was partially supported by an American Heart Association Grant in Aid and UTHSCSA Neurosurgery Stimulation Fund to Yuchuan Ding.