
Abstract
The calpain family of proteases is causally linked to postischemic neurodegeneration. However, the precise mechanisms by which calpains contribute to postischemic neuronal death have not been fully elucidated. This review outlines the key features of the calpain system, and the evidence for its causal role in postischemic neuronal pathology. Furthermore, the consequences of specific calpain substrate cleavage at various subcellular locations are explored. Calpain substrates within synapses, plasma membrane, endoplasmic reticulum, lysosomes, mitochondria, and the nucleus, as well as the overall effect of postischemic calpain activity on calcium regulation and cell death signaling are considered. Finally, potential pathways for calpain-mediated neurodegeneration are outlined in an effort to guide future studies aimed at understanding the downstream pathology of postischemic calpain activity and identifying optimal therapeutic strategies.
Introduction
Since their discovery over 40 years ago, members of the calpain family of calcium-regulated cysteine proteases have been linked to a wide range of pathologic conditions including postischemic neuronal cell death. However, much remains unknown about the precise mechanism of calpain-mediated neuronal injury and the specific calpain substrates that play essential roles in postischemic neuronal death. This review presents an organized approach to explaining the potential roles of different calpain isoforms and substrates at key subcellular locations during postischemic neurodegeneration.
Overview of the Calpain—Calpastatin System
The calpain family of proteases was first established by the discovery of what is now known as μ-calpain in 1964 (Guroff, 1964). The cDNA for this protein was subsequently isolated in the mid-1980s (Ohno et al, 1984), and based on sequence homology, there are now 15 identified calpain family members within the human genome. The majority of these have been identified only as mRNA, and several are thought to be tissue specific (Goll et al, 2003). Only calpain catalytic subunit isoforms 1, 2, 3, 5, 10, and two small regulatory subunit isoforms have been identified in the brain. In addition to the various protease isoforms, the calpain system includes a single endogenous inhibitor, calpastatin. The various calpain isoforms are thought to have a common substrate profile. Despite multiple attempts at substrate sequence analysis (Tompa et al, 2004; Cuerrier et al, 2005), no definitive method exists to predict whether a given compound is a calpain substrate, or, if it is a substrate, to identify the cleavage site. Furthermore, there is evidence that cleavage can be regulated by modulation of secondary recognition sequences adjacent to the actual cleavage site (Wang and Yuen, 1999).
The most abundant and best-characterized brain calpains are the two major isoforms μ- and m-calpain. Both these proteases exist as heterodimers, each with a distinct 80-kDa catalytic subunit encoded by the CAPN1 gene on chromosome 11 or the CAPN2 gene on chromosome 1, respectively. Henceforth, the individual catalytic subunits will be referred to as calpains 1 and 2, whereas the heterodimeric peptides they form with their common regulatory subunit will be termed μ- and m-calpain, respectively. The 80-kDa calpain 1 and 2 subunits share approximately 50% sequence homology, and amino-acid sequence analysis of both predicts four functional domains (Ohno et al, 1984). The domain maps of calpains 1 and 2 are shown in Figures 1A and 1B, respectively. Calpain 2, the first to be crystallized (Hosfield et al, 1999), has an anchor domain at the N-terminus of the protein that undergoes autolytic cleavage, likely through an intermolecular mechanism. The catalytic site is present in domain II. The precise function of domain III is unknown, but it is likely important for phosopholipid and calcium binding (Tompa et al, 2001) as well as other electrostatic interactions (Strobl et al, 2000). Domain IV is the calcium-binding domain, which contains five EF-hand motifs. In the absence of calcium, the catalytic triad consisting of Cys105 in domain I and His262 and Asn286 in domain II is separated, indicating that a conformational change is required for activation (Hosfield et al, 1999). The crystal structure of a chimeric peptide containing 85% of the sequence of calpain 1 essentially mimics the structure of calpain 2 (Pal et al, 2003). The important distinctions between the two structures are that in calpain 1 the separation of the catalytic residues is less dramatic, and the calcium-binding region of domain IV is more flexible. These findings could explain the primary biochemical distinction between μ- and m-calpain, namely the concentration of calcium required for in vitro activation. μ-Calpain requires 3 to 50 μmol/L Ca2+ for half-maximal activity, whereas m-calpain requires higher levels, 400 to 800 μmol/L (Goll et al, 2003). Both major calpains share a common regulatory subunit, known as calpain small subunit (CSS)1 (also referred to as calpain 4; gene name, CAPNS1). This subunit consists of two domains (Figures 1A and 1B): the hydrophobic N-terminal domain V is glycine-rich and the C-terminal domain VI, like domain IV of the catalytic subunits, is a calcium-binding region containing five EF-hand motifs (Hosfield et al, 1999). More recently, one group has described a second small subunit, CSS2 (gene name, CAPNS2), which has 73% sequence homology with CSS1, but which binds calpains 1 and 2 with less affinity (Schád et al, 2002). They further described differential distribution of CSS1 and CSS2 within rat brain, with CSS1 present in dendrites and CSS2 in axons (Friedrich et al, 2004). However, given that knockout of CSS1 is lethal despite the presumed continued expression of CSS2, it does not appear that the more recently identified subunit is able to functionally replace CSS1.
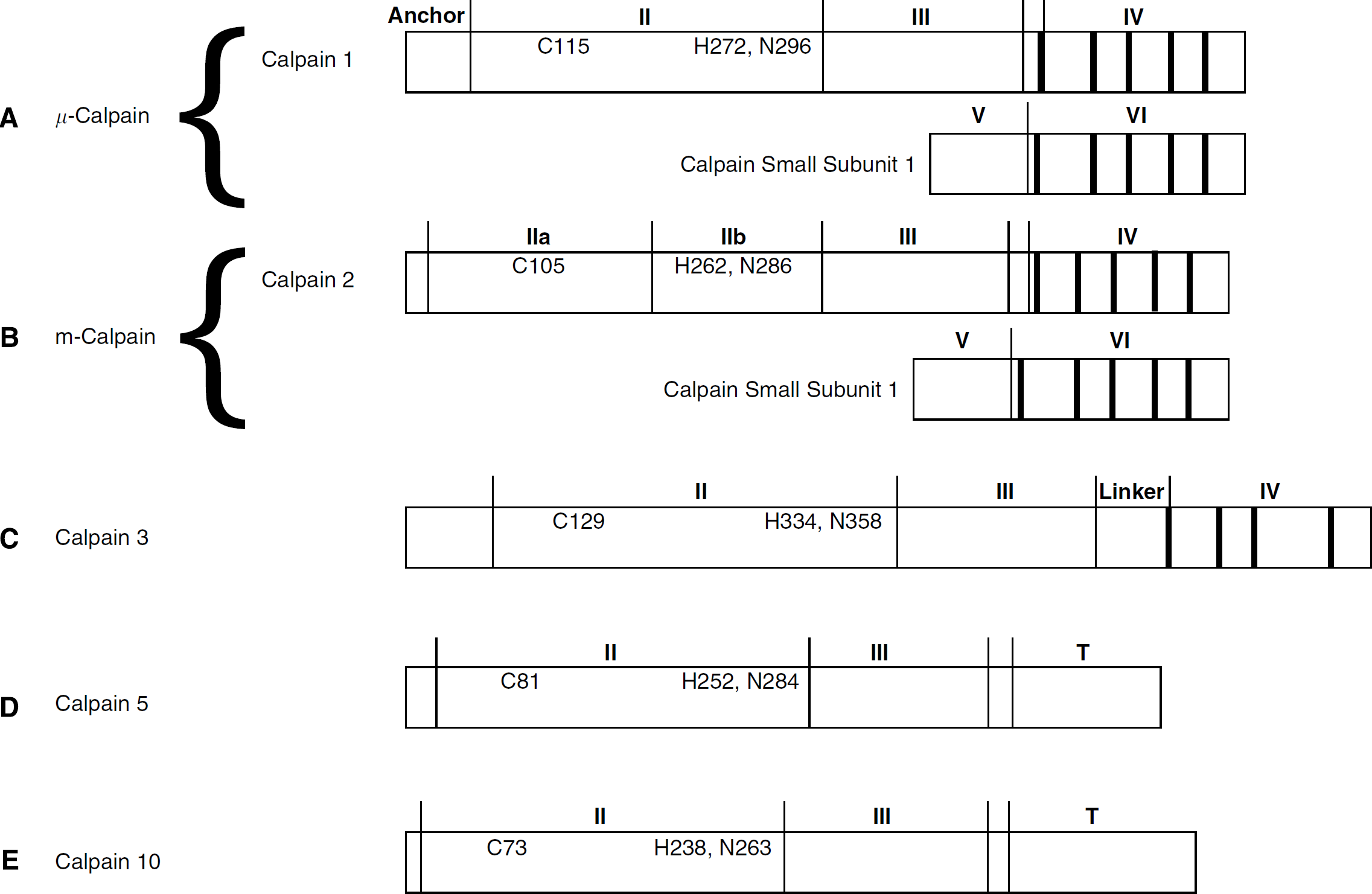
Domain structure of brain calpain isoforms. Schematic representations based on crystallographic data for μ-calpain (
Three other calpain family members have been identified in neural tissue. mRNA for calpain 3, previously believed to be skeletal muscle specific, has been identified in brain homogenates (Herasse et al, 1999). Antibodies raised from synthetic calpain 3 peptide sequences have been used to show calpain 3 protein in astrocytes (König et al, 2003) and a neuronal cell line (Marcilhac et al, 2006). Unlike the heterodimeric μ- and m-calpain, calpain 3 is a 94-kDa monomeric protease similar to the 80-kDa calpain 1 and 2 catalytic subunits (Figure 1C). mRNA for calpain 5 has been identified in human and rat brain. Like calpain 3, calpain 5 is a monomeric protein; however, it lacks domain IV, which is replaced by a unique T domain, the function of which is unknown (Figure 1D). The substitution of the T domain for domain IV is also seen in calpain 10 (Figure 1E), which has been identified in homogenates of rat retina, heart, skeletal muscle, and brain using an antibody generated from a synthetic peptide (Ma et al, 2001). The Caenorhabditis elegans homolog of calpain 5 is necessary for neurodegeneration in that species (Syntichaki et al, 2002), whereas the role of calpain 5 in other models of brain injury is not known. Similarly, calpains 3 and 10 may have roles in apoptotic cell death (Marcilhac et al, 2006) and mitochondrial dysfunction (Arrington et al, 2006), respectively, whereas their roles in brain physiology and pathology have yet to be determined.
Calpastatin is the only known protease inhibitor that appears to be completely specific to calpains. Both initial activation and subsequent calpain activity are inhibited by calpastatin, and because of its repeated domain structure, each calpastatin molecule is able to inhibit four calpains (Maki et al, 1987). Calpastatin alone does not bind calcium, whereas it binds to calpain only in the presence of calcium (Takano et al, 1999). Calpain alone possesses the ability to cleave calpastatin, but the proteolytic fragments retain their inhibitory function (DeMartino et al, 1988). One study has also suggested that the relative inhibitory effect of calpastatin differs between the two major isoforms, depending on its phosphorylation state. Dephosphorylated calpastatin has a 3- to 4-fold lower Ki against μ-calpain, whereas phosphorylated calpastatin has a 3- to 4-fold lower Ki against m-calpain (Salamino et al, 1994a). The other calpain isoforms are also believed to be inhibited by calpastatin, but detailed studies of the inhibitory effects have not been reported.
Physiologic Activity
The role of calpains in physiologic processes has been extensively reviewed (Goll et al, 2003) and encompasses functions including cell cycle regulation, differentiation, cell migration, adhesion, and signal transduction. A similarly wide range of physiologic functions has been attributed to calpain within the brain and neuronal culture systems. Calpain has been implicated in the differentiation of neuronal cell lines (Grynspan et al, 1997), neurite outgrowth in hippocampal neuronal culture (Song et al, 1994), as well as synaptic remodeling and long-term potentiation (Tomimatsu et al, 2002).
Through the use of transgenic mice and RNA interference (RNAi), there is an increasing literature concerning the isoform specificity of some physiologic functions of calpains. Perhaps the most convincing evidence for a functional difference between μ- and m-calpain is the fact that mice with a knockout of the calpain 1 catalytic subunit have been reported to have defects in platelet function (Azam et al, 2001) and to have normal long-term potentiation (Grammer et al, 2005), whereas knockout of calpain 2 (Dutt et al, 2006) or the common regulatory subunit (Zimmerman et al, 2000; Arthur et al, 2000) is embryonic lethal. Both calpain-3 and calpain-10 knockouts are viable, but the effects of these gene deletions on brain physiology and pathology have not been reported.
Studies using antisense oligonucleotides or RNAi have similarly shown isoform-specific physiologic effects. μ-Calpain has a demonstrated role in muscle cell differentiation (Moyen et al, 2004) and cell motility and migration (Satish et al, 2005; Wu et al, 2006). m-Calpain is involved in myoblast fusion (Balcerzak et al, 1995), membrane protrusion (Franco et al, 2004), and chromosome alignment during mitosis (Honda et al, 2004). Antisense oligonucleotides targeting the calpain 1 catalytic subunit reduced the effects of
Localization
The localization of calpain within the central nervous system has been repeatedly investigated. Immunohistochemical results have varied widely, with most studies of μ-calpain agreeing that it is expressed in neurons, and some noting glial expression as well. Specific brain regions identified include cerebellar Purkinje cells (Siman et al, 1985; Hamakubo et al, 1986), spinal cord nuclei (Siman et al, 1985; Hamakubo et al, 1986; Fukuda et al, 1990), cortex (Perlmutter et al, 1990), regions of diencephalon (Siman et al, 1985; Perlmutter et al, 1990), caudate nucleus (Siman et al, 1985; Perlmutter et al, 1990), and both granular and pyramidal neurons within the hippocampus (Siman et al, 1985; Hamakubo et al, 1986; Perlmutter et al, 1988; Fukuda et al, 1990). One immunohistochemical study specifically examining m-calpain noted that its expression was limited largely to glia (Hamakubo et al, 1986), whereas another observed m-calpain in hippocampal interneurons (Fukuda et al, 1990). Variable results such as these are common among calpain localization studies: in another example, Siman et al (1985) identified μ-calpain only in the hippocampal dentate gyrus, whereas Hamakubo et al (1986) and Fukuda et al (1990) reported that it is present in pyramidal cells throughout the hippocampus. Furthermore, Hamakubo et al (1986) observed only m-calpain in glial cells, whereas Perlmutter et al (1990) observed μ-calpain in glia. The major drawback of these studies, and undoubtedly the source of their variable results, is the fact that each used a different antibody. Although the isoform specificity of these antibodies was established in western blots, it is not clear as to what level of cross-reactivity each might have for other calpain isoforms when used in immuno-histochemical studies.
To avoid this problem, more recent studies have examined mRNA, rather than protein, expression. The most definitive of these used a combination of reverse transcriptase-polymerase chain reaction and in situ hybridization to examine the relative abundance and localization of mRNAs for calpains 1 and 2 and calpastatin (Li et al, 1996). Expression of calpain 2 mRNA was found to be 15-fold higher than calpain 1 in whole-brain homogenates. Calpastatin mRNA expression was three-fold higher than calpain 1. Calpain 1 and calpastatin message were observed to be diffused throughout the entire brain in both neurons and glia, whereas message for calpain 2 localized to distinct neuronal populations, including all cornu ammonis regions of hippocampus, cortical pyramidal neurons, and cerebellar Purkinje cells.
Less well studied is the subcellular localization of various components of the calpain system. Early immunoelectron microscopy studies were able to identify μ-calpain throughout neuronal and glial cytoplasm, typically associated with cytoskeletal elements. In neurons, staining extended into axons and dendrites, and, more specifically, was associated with dendritic spines and postsynaptic densities (Perlmutter et al, 1988). As a result of this study and of rough localization claims in earlier immunohistochemistry studies, calpain has typically been considered a ‘cytoplasmic’ protein. More recent immunoelectron microscopic studies have also placed calpain in the cytoplasm, but associated with the membranes of the endoplasmic reticulum (ER) and Golgi apparatus (Hood et al, 2003). Calpains' involvement in nuclear processes such as mitosis (Honda et al, 2004), however, suggests that calpain at least possesses the ability to translocate to the nucleus. In fact, fluorescein-tagged μ-calpain has been shown to be selectively transported to the nucleus under conditions known to allow energy-dependent nuclear transport of proteins (Mellgren and Lu, 1994), both μ- and m-calpain have been identified in the nuclear fraction of rabbit hippocampal homogenates (Ostwald et al, 1994), and m-calpain has been observed in cerebellar granule cell nuclei by immunohistochemistry and subcellular fractionation (Tremper-Wells and Vallano, 2005). Additionally, a recent study showed the presence of μ-calpain within isolated mitochondria (Garcia et al, 2005). Further studies have localized μ-calpain specifically to the mitochondrial intermembrane space (Cao et al, 2007). Calpain 10 has also been localized to mitochondria (Arrington et al, 2006), whereas calpain 3 is present in the cytoplasm and nucleus (Marcilhac et al, 2006). The subcellular localization of calpain 5 within the central nervous system has not been reported.
In addition to the localization of calpain under baseline conditions, there have been numerous studies of calpain translocation throughout the cell. Activation of calpain is associated with translocation to the plasma membrane in neutrophils (Pontremoli et al, 1989) and platelets (Ariyoshi et al, 1993). The importance of calpain translocation in the brain is not clear, and there is some evidence that calpain may be present at the membrane before any excitotoxic insult, and therefore no translocation is necessary before calpain activation (Hewitt et al, 1998). Given the similar substrate profiles of the two major calpain isoforms, it is likely that localization and translocation within the cell play a key role in differentiating their physiologic roles, and may impact their function in pathology as well.
Role of Calpain in Postischemic Neurodegeneration
Several forms of evidence have emerged since the early 1990s to support a role for calpain activity in acute neural injury. Calpain activation and activity occur in a spatial and temporal pattern that suggests a possible causal role for calpain in postischemic neurodegeneration (Saido et al, 1993; Roberts-Lewis et al, 1994; Neumar et al, 1996, 2001; Bartus et al, 1998). This evidence is bolstered by a series of studies showing that neurodegeneration is prevented by treatment with calpain inhibitors before (Lee et al, 1991; Rami and Krieglstein, 1993; Hong et al, 1994; Yokota et al, 1999) or after an ischemic insult (Bartus et al, 1994a, 1994b; Li et al, 1998; Markgraf et al, 1998).
Four major classes of calpain inhibitors have been used in in vitro and in vivo models of ischemic injury. There are several factors that can limit the utility of these inhibitor studies, including specificity, cell permeability, and blood—brain barrier penetration (for review, see Wang and Yuen, 1999; Donkor, 2000). The first class, the peptide aldehydes, bind reversibly to the active site and have poor specificity—in addition to calpain, these compounds can inhibit plasmin, trypsin, papain, and especially cathepsins. Early peptide aldehydes also had poor cell permeability, but this has been improved in later compounds such as the commonly used MDL28170. The newer peptide aldehyde SJA6017 is an even more potent inhibitor of calpains than MDL28170, but it continues to inhibit cathepsins as well (Inoue et al, 2003). Although it has been effective in treating traumatic brain injury (Kupina et al, 2001) and hypoxia in cardiac myocytes (Aki et al, 2002), use of SJA6017 in models of brain ischemia has not been reported. Another group of inhibitors, the epoxysuccinyl peptides, bind irreversibly to the calpain active site and have even better cell permeability. While they have been neuroprotective in in vitro excitotoxic models (Brorson et al, 1995; Rami et al, 1997), these compounds also inhibit other cysteine proteases in addition to calpain. The third major class of calpain inhibitors, the ketoamides, has several advantages over previous compounds, including high cell permeability and water solubility. However, early compounds were effective only after direct application to the brain (Bartus et al, 1994a) or intraarterial administration (Bartus et al, 1994b). Newer ketoamides, including A705239, have high oral bioavailability (Lubisch et al, 2003). Although they still inhibit cathepsin B, these drugs have no crossinhibition of either papain or caspase-3. A705239 is neuroprotective after traumatic brain injury (Lubisch et al, 2003) and preserves myocardial function after cardiac ischemia (Neuhof et al, 2003; Trumbeckaite et al, 2003). The development of more specific calpain inhibitors has been further limited by the fact that formation of the calpain catalytic site only occurs once the protease has been activated (Hosfield et al, 1999). As a result, it is difficult to design active site inhibitors because the active form cannot be crystallized and therefore the exact structure of the catalytic site is unknown. An alternate approach has been to target regions outside the catalytic site. This approach is exemplified by the fourth class of inhibitors, α-mercaptoacrylic acids such as PD150606. This compound is highly calpain specific and binds outside the active site (Wang et al, 1996). The only reported in vivo use of PD150606 required intracerebroventricular infusion of the drug, limiting its clinical applicability (Farkas et al, 2004).
To address the problems inherent in synthetic calpain inhibitors, an attempt was made to create a fusion protein consisting of the endogenous calpain inhibitor calpastatin coupled to the HIV Tat transducing protein. Although the Tat—calpastatin fusion product was able to inhibit calpain in cell-free assays, and was readily taken up into neurons, it remained encapsulated in endosomes and displayed no inhibitory activity in the cell (Sengoku et al, 2004). Even if such an approach were to be successful, calpastatin, like all the other available inhibitors, is not isoform specific (Saito and Nixon, 1993). As a result, most studies implicating calpain in the pathology of postischemic neurodegeneration are unable to identify the specific isoform involved; therefore, henceforth, the generic term ‘calpain’ will be used in cases where a specific isoform has not been identified.
The limitations of these inhibitory strategies have more recently been overcome through the use of genetic techniques. Twenty-fold overexpression of calpastatin in human SH-SY5Y neuroblastoma cells was protective against low-dose staurosporine injury, but this effect was lost with more substantial injuries (Neumar et al, 2003). Subsequently, two papers have described the effects of acute brain-injury models in mice with neuron-specific overexpression of calpastatin. In both cases, the consequent reduction in calpain activity was associated with neuroprotection in a kainic acid-based excitotoxicity model (Takano et al, 2005; Higuchi et al, 2005). These last studies have definitively established the role of calpain in mediating acute brain injury. What remains unknown is the precise calpain isoform or isoforms responsible for postischemic pathologic calpain activity. One study using antisense oligonucleotides to reduce μ-calpain expression in organotypic hippocampal slice culture has shown protection against excitotoxicity (Bednarski et al, 1995). More recently, RNAi-mediated knockdown of μ-calpain, but not m-calpain, was protective in an in vitro model of apoptosis-inducing factor (AIF)-mediated cell death (Cao et al, 2007). The in vivo effects of isoform-specific knockdown on brain injury have not been investigated. The importance of identifying the isoform responsible for pathologic calpain activity is highlighted by the differing physiologic roles discussed above. This is particularly important because m-calpain appears to be essential for normal mitosis. Additionally, given the potentially different subcellular localization of the major brain calpain isoforms, identifying a single isoform responsible for pathologic activity may allow one to narrow down the number of calpain substrates that play a key role in neuronal cell death. This is perhaps the largest unanswered question in the study of postischemic calpain proteolysis, and the subject of the remainder of this review—by what mechanisms do calpains cause neuronal death?
Subcellular Consequences of Pathologic Calpain Activity
Given the large number and wide range of known calpain substrates, it has been difficult to develop a unified model of how calpain proteolysis leads to eventual cell death. In an effort to develop such a model, this paper will take two approaches for categorizing and discussing calpain substrate cleavage. The first of these is to consider calpain substrates by the subcellular location. The pathology of ischemic neuronal injury involves alterations in the function of multiple cellular components, including the synapse, plasma membrane, ER, mitochondria, lysosomes, and nucleus. All these neuronal regions or organelles contain calpain substrates, and, therefore, a systematic consideration of the subcellular localization of pathologic calpain activity is one method by which one can understand the mechanism of calpain-mediated cell death. Regional differences in calcium fluxes within neurons during ischemia and reperfusion further support this theoretical approach.
Calpain substrates also exist across a wide range of functional categories. Three important categories include postsynaptic structural proteins (Table 1), calcium regulatory proteins (Table 2), and signaling proteins (Table 3). Calpains require elevated calcium concentrations not only for initial activation but also for subsequent activity, although activity can be sustained with a lower degree of elevation (Goll et al, 2003). As a result, it makes sense that calpain itself may play some role in regulating calcium homeostasis. In fact, most neuronal calcium regulatory proteins are known calpain substrates. A second set of calpain substrates can be defined as those contributing to normal cell structure. Calpain cleavage of this set of targets leads directly to cellular breakdown that is a hallmark of postischemic neuronal death. However, it remains to be determined if cytoskeletal proteolysis is causally related to cell death or primarily a postmortem phenomenon. In addition to directly damaging the cell structure, calpains also engage in complex proteolytic signaling cascades. This final group of calpain substrates consists of other proteins that undergo changes in function, rather than simply loss of function, in response to calpain cleavage. As we consider each subcellular location, it will be important to evaluate the contribution each of these three categories of substrates makes toward eventual cell death.
Postsynaptic structural proteins cleaved by calpain

Abbreviations: AMPA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; MAP, microtubule-associated protein; NMDA, N-methyl-
Calcium regulatory proteins cleaved by calpain
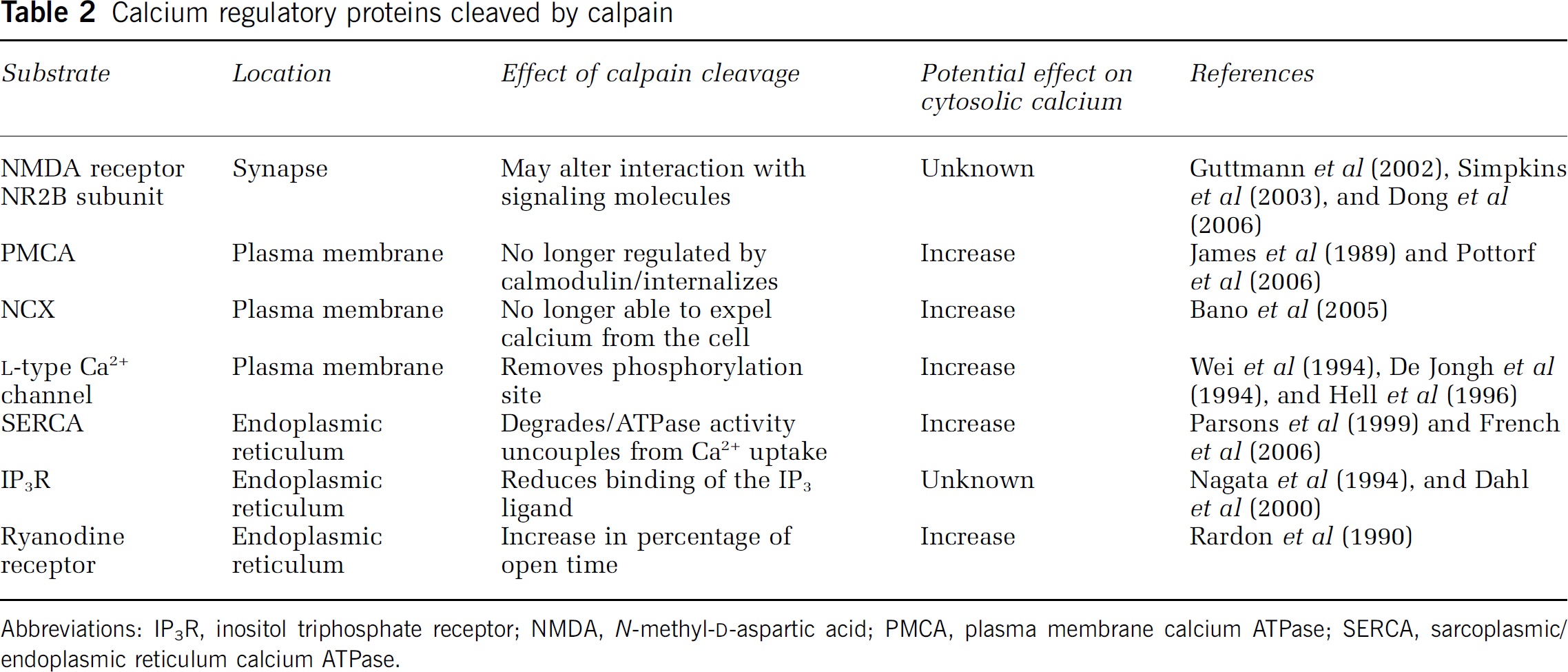
Abbreviations: IP3R, inositol triphosphate receptor; NMDA, N-methyl-
Signaling cascades influenced by calpain
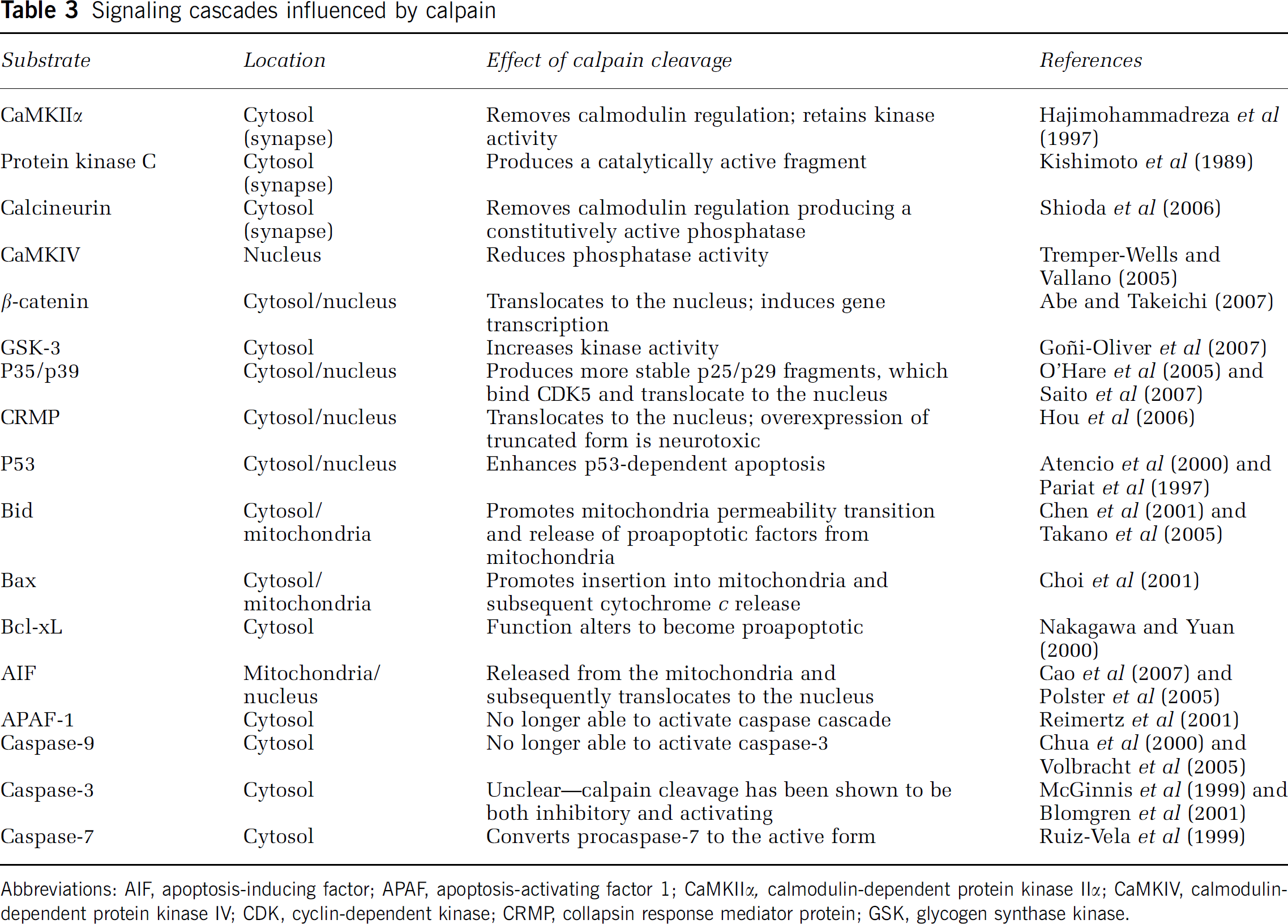
Abbreviations: AIF, apoptosis-inducing factor; APAF, apoptosis-activating factor 1; CaMKIIα, calmodulin-dependent protein kinase IIα; CaMKIV, calmodulin-dependent protein kinase IV; CDK, cyclin-dependent kinase; CRMP, collapsin response mediator protein; GSK, glycogen synthase kinase.
Plasma Membrane and Synapse
A major component of the postischemic injury cascade in the brain is the membrane depolarization that occurs after ATP depletion (for review, see Neumar, 2000). This membrane depolarization results in the opening of voltage-gated sodium and calcium channels and in the release of excitatory neurotransmitters, such as glutamate. Transmitter release further alters ion homeostasis as it results in activation of calcium-permeable NMDA receptors and sodium-permeable AMPA receptors. Resulting loss of the normal Na gradient then leads to reversal of the Na+/Ca2+ exchanger. Although these events are likely sufficient to activate calpain, the initial change in ion balance is reversible within minutes after a mild or moderate ischemic insult. However, there is a secondary disruption of ion homeostasis that occurs in many neurons between 4 and 48 h after reperfusion. This further disruption of calcium regulatory processes is necessary to produce the delayed calpain activation that potentially contributes to delayed postischemic neurodegeneration. Calpain itself may contribute to the sustained or secondary calcium overload by cleaving and altering the function of many of these proteins during the recovery period.
Plasma Membrane: The most obvious effects of calpain activity on calcium regulation come through its cleavage of
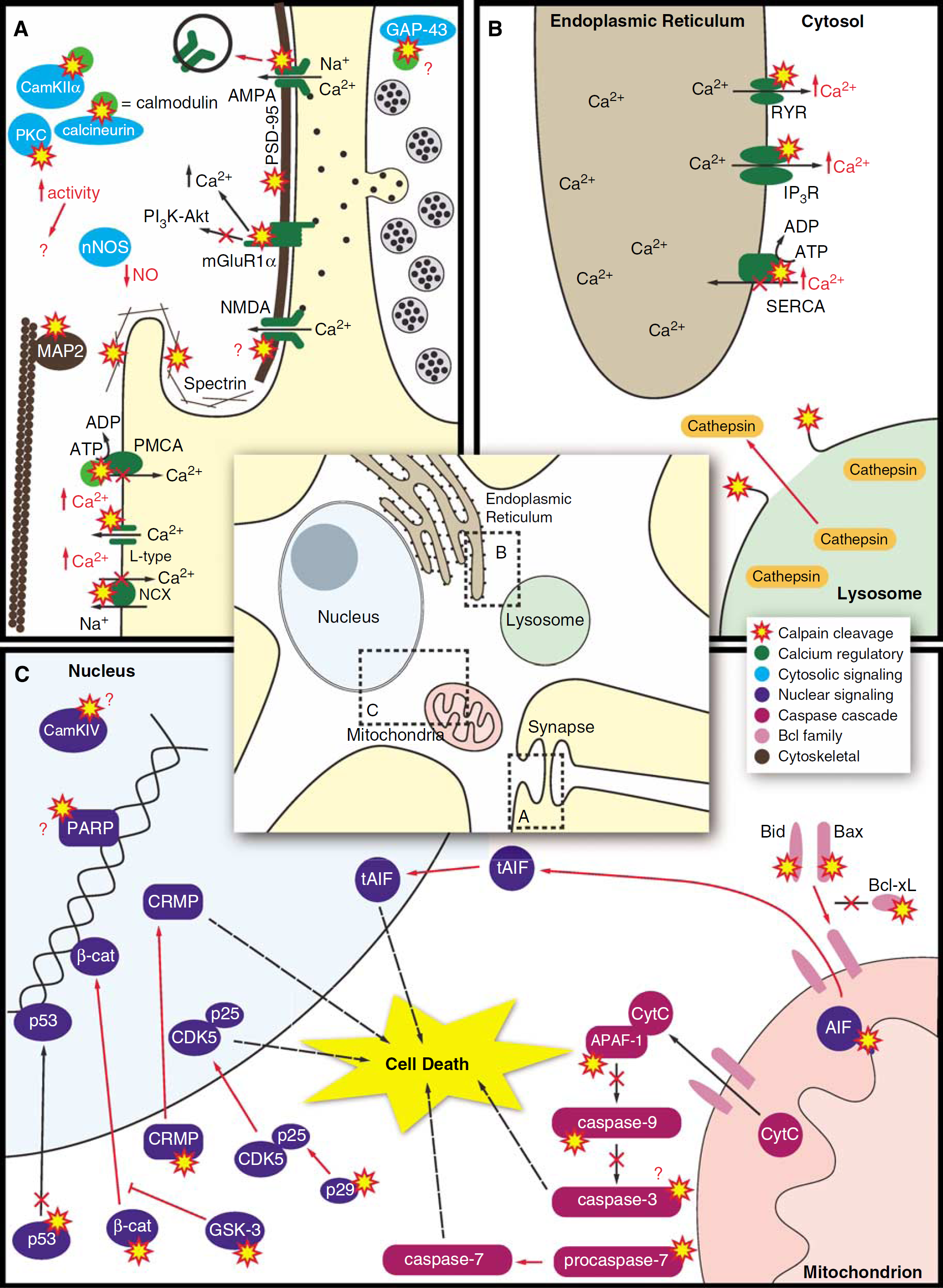
Subcellular localization of calpain pathology in postischemic neurons. Postischemic calpain activity occurs in a wide range of subcellular regions including the synapse and plasma membrane (
The plasma membrane calcium ATPase (PMCA) has long been known to be a calpain substrate in erythrocytes, where truncation results in an active form of the protein that is no longer regulated by calmodulin (James et al, 1989). However, at least one study has shown that calpain proteolysis of erythrocyte PMCA in situ actually causes a decrease in function (Salamino et al, 1994b). More recently, glutamate excitotoxicity has been shown to result in internalization of PMCA in hippocampal neurons, an effect that was blocked by calpain inhibition (Pottorf et al, 2006). This result is consistent with studies showing loss of PMCA activity (Oguro et al, 1995) and expression (Lehotský et al, 1999) in postischemic gerbil hippocampus. The result of calpain-mediated PMCA truncation therefore appears to parallel the effects seen for calpain cleavage of
Synapse: The manner in which calpain activity may alter calcium regulation through cleavage of neurotransmitter receptors is less straightforward (Figure 2A). Classic NMDA-type glutamate receptors are heteromeric proteins composed of two NR1 subunits and two NR2 subunits, selected from subtypes NR2A, NR2B, NR2C, and NR2D. Of these, NR2A, -2B, and -2C are known to undergo C-terminal proteolysis by calpain in vitro (Guttmann et al, 2002), a truncation that does not appear to prevent any aspects of receptor function. However, only the NR2B subunit has been (Xu et al, 2007) shown to be cleaved by calpain in mature neurons (Dong et al, 2006). This cleavage occurs both in vivo after transient bilateral carotid artery occlusion and in vitro after excitotoxic insults in hippocampal neurons (Simpkins et al, 2003). The cleaved receptor remains on the cell surface, and, although it has not been shown to be active in neurons, it is predicted to be so based on the cleavage sites. Separate published reports have shown either a reduction in NMDA-mediated transmission (Wu et al, 2005) or essentially no change in electrophysiologic properties (Guttmann et al, 2001) after calpain proteolysis. What should be altered is the ability of the truncated C-terminus to interact with intracellular signaling molecules. The downstream effects of this modification on signaling and calcium regulation are unknown.
Activation of NMDA receptors is also associated with calpain-mediated cleavage of a member of the metabotropic family of glutamate receptors. The C-terminus of the mGluR1α G-protein-coupled receptor is cleaved by calpain after NMDA exposure (Xu et al, 2007). The truncated receptor retains its ability to signal for the release of calcium from intracellular stores, but is unable to activate the neuroprotective PI3 kinase—Akt signaling pathway. The calpain-cleaved mGluR1α is therefore able to contribute to cytosolic calcium overload without any balancing neuroprotective effect.
Calpain also cleaves the GluR1, GluR2, and GluR3 subunits of AMPA-type ionotropic glutamate receptors (Bi et al, 2000). Again, calpain proteolysis produces a C-terminal truncation of the receptor, an effect that is seen in vitro and after NMDA excitotoxicity in hippocampal slice cultures (Bi et al, 1998). The effects of calpain cleavage are clearer for AMPA receptors than for NMDA receptors, with studies suggesting that calpain activation leads to both a loss of total GluR1–3 protein (Jourdi et al, 2005) and their dissociation from postsynaptic densities (Lu et al, 2000b). The overall conclusion from these studies appears to be that calpain cleavage reduces AMPA-mediated currents and increases turnover of AMPA receptors. This is particularly interesting in light of studies showing a decrease in expression of mRNA for the calcium-impermeable GluR2 subunit after ischemia, suggesting that replacement of AMPA receptors after ischemia would be biased toward calcium-permeable isoforms (Pellegrini-Giampietro et al, 1997). This concept is supported by the fact that overexpression of a Ca2+-impermeable form of the receptor rescues neurons from ischemic damage (Liu et al, 2004). Conversely, it has recently been shown that the calpain-mediated downregulation of AMPA receptors acts primarily on the GluR1 subunit, leaving GluR2 at the cell surface both in cortical neuron culture and in rat brain (Yuen et al, 2007). It is not clear what effect this sparing of GluR2 subunits might have on potential changes in Ca2+ permeability associated with AMPA receptor turnover.
In addition to direct effects on neurotransmitter receptors, postischemic calpain activation may alter synapse structure and function through cleavage of postsynaptic cytoskeletal elements (summarized in Table 1 and Figure 2A). Postsynaptic density protein 95 ordinarily binds numerous receptors and protects NR2A from being cleaved by calpain (Dong et al, 2004). However, postsynaptic density protein 95 itself is cleaved by calpain in calcium-treated rat brain sections and NMDA-exposed hippocampal slice culture (Lu et al, 2000a), which may modify the anchoring of NMDA receptors (Vinade et al, 2001). Calpain also cleaves proteins associated with the stabilization of AMPA receptors, such as synapse associated protein-97 (SAP-97) and glutamate receptor-activating protein (Jourdi et al, 2005), which may have an additive effect with direct calpain cleavage of AMPA subunits on receptor turnover.
The structural changes that occur in synapses after an ischemic insult extend beyond tethering of receptors. Rapid changes in dendritic spine density have been observed within hours of in vitro excitotoxicity, where calpain appears to be involved in synaptic remodeling (Faddis et al, 1997). These changes in neuronal structure likely involve cleavage of cytoskeletal calpain substrates such as αII-spectrin and microtubule-associated protein II (MAP2).
Spectrin is the major cytoskeletal protein component of the cell membrane (Czogalla and Sikorski, 2005). Intact spectrin is a 280 kDa protein, calpain cleavage of which produces unique 150 and 145 kDa fragments. Antibodies specific to these fragments provide a means to identify calpain activity, both through Western blotting of tissue lysates and through immunohistochemistry. Studies in postischemic gerbil hippocampus have shown an initial rapid rise in calpain-cleaved spectrin that occurs within 15 mins of reperfusion and which is then followed by a secondary accumulation of spectrin breakdown product between 4 and 24 h after injury (Saido et al, 1993; Roberts-Lewis et al, 1994). Transient forebrain ischemia in rats produces a similar pattern, with a peak in spectrin breakdown products observed at 1 h after injury that subsides only to be followed by a second peak beginning between 24 and 48 h in those cornu ammonis 1 hippocampal neurons that subsequently degenerate (Neumar et al, 2001). Spectrin proteolysis is, in fact, a hallmark of postischemic brain injury to the degree that the appearance of spectrin breakdown products in cerebrospinal fluid can be used as a biomarker of ischemia (Siman et al, 2004). Given the interactions between spectrin and many other membrane proteins such as ankyrin, actin, calmodulin, and microtubules (Bennett, 1990), it is likely that postischemic spectrin cleavage by calpain has substantial effects on membrane structure and function.
Microtubule-associated protein II serves to stabilize microtubules and regulate the microtubule network and transport system in neuronal dendrites (Dehmelt and Halpain, 2005). Microtubule-associated protein II is susceptible to calpain-mediated cleavage, as in vivo excitotoxicity leads to calpain-mediated loss of MAP2 immunoreactivity (Siman and Noszek, 1988). Similar degradation of MAP2 occurs after oxygen—glucose deprivation in hippocampal slice culture (Buddle et al, 2003) and in in vivo focal (Pettigrew et al, 1996) and global ischemia (Ziemka-Nałecz et al, 2003). This degradation of MAP2 is preceded by calpain activation, and is prevented by calpain inhibition (Inuzuka et al, 1990). Taken together, evidence for calpain-mediated cleavage of spectrin and MAP2 strongly suggests an important role of calpain in postischemic degradation of the neuronal cytoskeleton and alterations in synaptic structure.
The final role of calpain at the plasma membrane and in the synapse is to proteolytically modify a number of signaling molecules, triggering downstream effects throughout the cell. Many of these molecules are regulated by calmodulin, and a common effect of calpain cleavage is to remove this regulation. Excitotoxicity in hippocampal neurons (Wu et al, 2004) and focal brain ischemia in mice (Shioda et al, 2006) induces calpain cleavage of the calmodulin-dependent phosphatase calcineurin, producing a constitutively active form. Calpain also produces an active form of type IIα calmodulin-dependent protein kinase and degrades neuronal nitric oxide synthase after both in vitro and in vivo administration of a variety of neurotoxins (Hajimohammadreza et al, 1997). The presynaptic growth-associated protein 43, which is involved in the regulation of neurotransmitter release, is cleaved by calpain, decreasing calmodulin binding (Zakharov et al, 2005). Although not regulated by calmodulin, protein kinase C is cleaved by calpain, again producing a catalytically active fragment (Kishimoto et al, 1989). The precise downstream effects of cleavage of these signaling molecules on postischemic neurodegeneration are not known.
Endoplasmic Reticulum
Although the influx of extracellular calcium is certainly an important component of postischemic injury, it is important to also consider ways in which intracellular calcium stores can contribute to alterations in ion homeostasis. As noted above, calpain has been shown to be associated with the membrane of the ER (Hood et al, 2003, 2004). It is therefore only natural to assume that it is able to cleave ER proteins, and, in fact, several calpain substrates have been identified in the membrane of the ER (Figure 2B).
The ryanodine receptor (RYR) and inositol triphosphate receptor (IP3R) are the two Ca2+ channels found in the membrane of the ER. The primary physiologic mechanism of RYR channel activation is thought to be increased cytosolic Ca2+ (Ca2+-induced Ca2+ release). Calpain-mediated proteolysis of cardiac RYR2 has been studied in detail. Digestion of cardiac junctional sarcoplasmic reticulum vesicles by m-calpain causes cleavage of the 400 kDa RYR2 isoform into 350 and 315 kDa intermediates followed by generation of a 150 kDa limit protein (Rardon et al, 1990). Electron microscopic analysis of sarcoplasmic reticulum vesicles after m-calpain proteolysis revealed a ‘shaved-off’ appearance of the cytoplasmic extension of the proteins. The functional effect of calpain-mediated proteolysis of cardiac RYR2 has been studied with the protein reconstituted in lipid bilayers. In these studies, calpain-mediated cleavage of RYR2 renders the channel unable to close in the presence of elevated cytosolic Ca2+ levels. Specifically, addition of calpain to the cis (cytosolic) chamber containing 1 μmol/L Ca2+ causes the RYR2 percentage open time to increase from 36% to >90% with no change in unitary channel conductance (Rardon et al, 1990). Ryanodine receptor 2 channels in a ‘locked open’ state are a potential mechanism for permanent ER Ca2+ depletion and loss of the ER's ability to buffer elevations in cytosolic Ca2+.
Published work evaluating the effect of brain ischemia on RYR is limited to studies showing decreased 3H-ryanodine binding in situ after permanent unilateral carotid artery occlusion in gerbils (Nozaki et al, 1997, 1999). Interpretation of these studies is limited because ryanodine binding is not significantly altered by calpain cleavage (Rardon et al, 1990). However, postischemic administration of the RYR channel antagonist dantrolene has been reported to be neuroprotective both in vitro (Wei and Perry, 1996) and in vivo (Nakayama et al, 2002), suggesting that increased Ca2+ efflux via RYR could contribute to sustained or secondary postischemic calpain activation and eventual neuronal death.
The IP3R is the second Ca2+ channel in the ER. Inositol triphosphate receptor channel opening is stimulated by binding of IP3, a second messenger generated when phospholipase C converts phospha tidylinositol bisphosphate (PIP2) to IP3 and diacylglycerol. There is also evidence that IP3R Ca2+ release is enhanced by increased intracellular Ca2+, similar to RYR. The Ca2+-dependent channel opening probability displays a bell-shaped curve with maximum probability of opening at 0.2 μmol/L free Ca2+, which is significantly lower than the RYR (Bezprozvanny et al, 1991). Fifty percent of brain ER Ca2+ can be released by IP3 (Ferris and Snyder, 1992), making the IP3R a major potential player in postischemic calcium overload.
Calpain-mediated proteolysis of IP3R1 from the rat cerebellum or striatum results in loss of intact protein (260 kDa) and generation of 200, 130, and 95 kDa (predominant) fragments detected by antibody raised against a peptide corresponding to the C-terminal domain (Igwe and Filla, 1997). This proteolysis of the IP3R resulted in decreased IP3 binding, suggesting that site-specific cleavage decreases the affinity of the remaining protein species for the IP3 ligand. A possible calpain-mediated reduction in ligand binding is consistent with studies of IP3R in postischemic brain. Reduced 3H-IP3 binding has been observed in cornu ammonis 1 (CA1) hippocampus after unilateral carotid occlusion in gerbils, an effect that was not associated with any decrease in IP3R immunoreactivity (Nagata et al, 1994). A more recent study of global ischemia in the rat produced similar results—a reduction in 3H-IP3 binding in CA1, but no change in receptor expression (Dahl et al, 2000). The loss of IP3 binding has also been shown functionally in postischemic brain tissue. Flash-frozen hippocampal slices taken from gerbils after unilateral carotid artery occlusion showed normal uptake of 45Ca2+, but failed to release that calcium when stimulated with IP3. Although this would seem to indicate that calpain cleavage of the receptor prevents evoked calcium release, it is possible that the proteolysis simply removes the ligand regulation of the channel. It is easy to imagine a model in which calpain-mediated loss of the ligand-binding domain of the IP3R leads to baseline calcium release from the ER, contributing to postischemic calcium overload.
In addition to its effects on the IP3R, calpain may also alter ER calcium release by affecting processing of IP3 itself. A major metabolic pathway that serves to limit the half-life of IP3 is its phosphorylation to form IP4 by a family of IP3 kinases (Irvine et al, 1986). The B isoform of the IP3 kinase (IP3-KB) is cleaved by calpain (Pattni et al, 2003). This cleavage separates the catalytic and membrane-anchoring domains of the molecule, releasing IP3-KB from the membrane of the ER. This could then prevent metabolism of IP3, allowing it to act longer on the IP3R, thereby potentiating calcium efflux from the ER.
The sarcoplasmic/ER Ca2+ ATPase (SERCA) is responsible for energy-dependent ER Ca2+ sequestration. Sarcoplasmic/ER Ca2+ ATPase has been shown to be susceptible to calpain proteolysis in vitro, but only when the ATPase is in its oxidized form. Although calpain-mediated degradation of SERCA has been repeatedly shown after ischemia—reperfusion injury in the heart (French et al, 2006), little data exist for the effects of ischemia on brain SERCA. One study has measured SERCA ATPase activity in brain microsomes after decapitation ischemia (Parsons et al, 1999). In this model, SERCA-mediated 45Ca2+ uptake was decreased after as little as a 10-min ischemia, but SERCA-specific ATP hydrolysis was unaffected. This apparent uncoupling of SERCA ATPase activity and Ca2+ uptake could be explained by modification of the SERCA molecule itself or increased ER membrane Ca2+ conductance via RYR and/or IP3R channels. One would not expect calpain-mediated proteolysis of SERCA in a model of decapitation ischemia because unoxidized SERCA is not susceptible to calpain-mediated proteolysis. Sarcoplasmic/ER Ca2+ ATPase function has not yet been examined in the reperfused brain where conditions favor SERCA oxidation. Still, loss of SERCA function because of calpain-mediated proteolysis, like calpain alteration of RYR and IP3R systems, is a potential mechanism of Ca2+ overload in postischemic neurons.
Mitochondria
Mitochondrial dysfunction has a well-established role in ischemic brain injury. Like cytosolic calcium overload, mitochondrial calcium overload is known to occur both immediately after brain ischemia and again at later time points (Zaidan and Sims, 1994). The initial postischemic calcium overload is accompanied by a burst of free radical production (Hall et al, 1993), an effect that can be inhibited with mitochondrial complex I blockers (Piantadosi and Zhang, 1996). Mitochondrial permeability transition has been implicated in models of both focal (Shiga et al, 1992) and global (Uchino et al, 1995) brain ischemia by the neuroprotective effects of cyclosporine A, a mitochondrial permeability transition inhibitor. Although such mitochondrial dysfunction is classically associated with apoptotic cell death and activation of the caspase family of proteases, a number of studies have identified a role of calpain in cell death associated with mitochondrial dysfunction. Calpain inhibitors have been shown to preserve mitochondrial function in cardiac tissue after ischemia—reperfusion injury (Neuhof et al, 2003; Trumbeckaite et al, 2003). This effect is likely independent of any inhibition of caspases, as the specific inhibitors used have been shown not to inhibit caspase-3 (Lubisch et al, 2003).
Apoptosis Regulatory Proteins: One way in which calpain may contribute to mitochondrial dysfunction is through cleavage of a number of proteins that regulate apoptosis (Figure 2C). This includes the Bcl2 family of proteins, several of which are calpain substrates. Calpain cleavage of Bax produces an 18 kDa fragment that leads to cytochrome c release in dopaminergic neurons exposed to mitochondrial toxins (Choi et al, 2001). Similar results have been observed for Bid, another proapoptotic Bcl2 family protein. Calpain-mediated cleavage and activation of Bid have been observed in rabbit heart after ischemia reperfusion injury (Chen et al, 2001) and in mouse brain after kainate excitotoxicity (Takano et al, 2005). Bid activation in this model was enhanced in mice deficient for the endogenous calpain inhibitor calpastatin. There is also evidence that calpain cleaves the loop region of the antiapoptotic protein Bcl-xL, converting it to a proapoptotic molecule (Nakagawa and Yuan, 2000). Calpain cleavage of these molecules leads to mitochondrial permeability transition, and release of death trigger molecules such as AIF and cytochrome c. Calpain is also thought to regulate apoptosis through degradation of the transcription factor p53 (Pariat et al, 1997), a finding that is supported by the fact that inhibition of calpains increases activated p53 and enhances p53-dependent apoptosis in tumor cell lines (Atencio et al, 2000).
Caspase-Dependent Programmed Cell Death: Calpain may also play an important role in apoptotic cell death through direct and indirect interactions with members of the caspase family of proteases (Figure 2C). Apoptosis protease-activating factor-1, which combines with cytochrome c to bind and activate caspase-9, is cleaved by calpain. The resulting loss of apoptosis protease-activating factor-1 was correlated with reduced activity of caspase-3-like proteases (Reimertz et al, 2001). Direct calpain truncation of caspase-9 also results in a loss of ability to activate caspase-3 (Chua et al, 2000). Conversely, calpain cleavage converts procaspase-7 to the active form, independent of the activity of other caspases (Ruiz-Vela et al, 1999). Conflicting results have been reported for caspase-3, calpain cleavage of which has been shown in separate studies to be inhibitory (McGinnis et al, 1999) or activating (Blomgren et al, 2001). Crosstalk between the calpain and caspase systems also occurs via calpastatin. Caspases have been shown to cleave calpastatin and reduce its inhibitory ability (Wang et al, 1998), setting up a complex interaction between the calpain and calpastatin systems. Although overexpression of calpastatin initially enhances caspase-3 activity, likely by preventing calpain-mediated degradation of caspase-9 and caspase-3, calpastatin itself is degraded by caspase-3, resulting in a secondary increase in calpain activity (Neumar et al, 2003).
Caspase-Independent Programmed Cell Death: In addition to promoting formation of the mitochondrial permeability transition pore, calpain may play a more direct role in the release of AIF. Calpain has recently been shown to cleave AIF, and to induce its release from mitochondria (Polster et al, 2005; Cao et al, 2007). This cleavage can occur both with exposure to exogenous calpain and when the only calpain present is that already within the mitochondria. Release of AIF from the mitochondria has also been observed in concert with activation of caspase-12, another calpain substrate (Nakagawa and Yuan, 2000), in models of ER stress-induced apoptosis in retinal ganglion cells (Sanges et al, 2006). Release of AIF in response to excitotoxic stimuli is suppressed by adenovirus-mediated expression of calpastatin (Volbracht et al, 2005; Cao et al, 2007), in transgenic mice overexpressing calpastatin (Takano et al, 2005), and after RNAi-mediated knockdown of μ-calpain in neurons (Cao et al, 2007). Therefore, although calpain appears to downregulate forms of caspase-mediated cell death, it may be simultaneously working to promote caspase-independent programmed cell death via an AIF-mediated mechanism.
Lysosomes
One proposed mechanism by which calpain activation may bring about necrotic destruction of the cell is through release of the hydrolytic enzymes typically sequestered within lysosomes, primarily the cathepsins (Figure 2B). This process, described by Tetsumori Yamashima as the ‘calpain—cathepsin cascade’, consists of calpain-mediated disruption of lysosomal membranes followed by diffusion of cathepsin B throughout the cytoplasm and nucleus. The evidence supporting this hypothesis in models of brain ischemia has recently been reviewed (Yamashima, 2004). Independent support for the interaction between calpains and cathepsins comes from work in C. elegans where loss of function in the proteases CLP-1 and TRA-3 (equivalent to calpains) as well as ASP-3 and ASP-4 (equivalent to cathepsins) is neuroprotective (Syntichaki et al, 2002). Combined loss of both calpain and cathepsin equivalent proteases did not have an additive neuroprotective effect, suggesting that the two protease families may induce neurodegeneration via a common sequential pathway.
Nucleus
Calpain directly cleaves nuclear proteins as well as causes the translocation of cytosolic proteins to the nucleus after proteolysis (Figure 2C). Within the nucleus of primary cerebellar granule neurons, calpain cleaves Ca2+/calmodulin-dependent protein kinase type IV (CaMKIV), leading to a reduction in phosphorylation of CaMKIV targets (Tremper-Wells and Vallano, 2005). The role of calpain-mediated CaMKIV downregulation in neurodegeneration has not been investigated. Calpain also cleaves the nuclear protein poly(ADP-ribose) polymerase-1 (PARP). Poly(ADP-ribose) polymerase-1 is normally a DNA repair enzyme that becomes overactivated after numerous toxic insults, leading instead to NAD and ATP depletion and eventual cell death (Pellicciari et al, 2004). Evidence for a role in ischemic brain injury includes findings that PARP becomes activated after excitotoxicity in cerebellar granule neurons (Cosi et al, 1994) and that PARP gene expression increases after global brain ischemia or kainate injection in gerbil and rat brains (Liu et al, 2000). Knockout of PARP reduces infarct size in a mouse model of focal cerebral ischemia (Eliasson et al, 1997). There is also evidence that PARP plays a key role in AIF-mediated programmed cell death (Yu et al, 2006). Although PARP is a known calpain substrate (McGinnis et al, 1999), it is again not clear what role, if any, calpain-mediated PARP cleavage plays in postischemic neurodegeneration.
Calpain also cleaves a number of cytosolic molecules that subsequently translocate to the nucleus. One such molecule is β-catenin, which undergoes calpain-mediated truncation in neurons exposed to glutamate (Abe and Takeichi, 2007). The truncated molecule then translocates to the nucleus where it induces gene transcription. The precise role of this gene regulation in postischemic neuronal death has not been examined. A related molecule in the Wnt-signaling pathway, glycogen synthase kinase 3, is also cleaved by calpain. Like many other calpain substrates, cleaved glycogen synthase kinase remains functional, and, in fact, displays an increase in kinase activity (Goñi-Oliver et al, 2007). Interestingly, glycogen synthase kinase 3 phosphorylation of β-catenin should prevent its translocation to the nucleus, further complicating the role calpain cleavage of these molecules may have in postischemic pathology.
More substantial evidence for a causal role in calpain-mediated cell death exists for cyclin-dependent kinase 5 (CDK5). Cyclin-dependent kinase 5 is unique in the CDK family in that it is primarily active in the central nervous system, where it is involved in regulating neuronal migration and synaptic function (Dhavan and Tsai, 2001). Activity of CDK5 kinase is regulated by the binding of one of its activator proteins, p35 or p39, which are cleaved by calpain to form to p25 or p29, respectively (Kusakawa et al, 2000; Patzke and Tsai, 2002). Accumulation of p25 has been shown after excitotoxicity, hypoxia, calcium overload, and ER stress in primary neuronal culture (Lee et al, 2000; Saito et al, 2007), as well as after middle cerebral artery occlusion in vivo (Nath et al, 2000). Similar results have been reported for p29 (Patzke and Tsai, 2002). Calpain-mediated truncation of p35 to p25 produces a more stable form of the protein, which is still able to activate CDK5. The CDK5–p25 complex translocates from the cytoplasm to the nucleus, where it appears to play an essential role in neuronal cell death (O'Hare et al, 2005; Saito et al, 2007).
The collapsin response mediator proteins (CRMPs) are another family of proteins with important roles in normal neuronal development that undergo calpain cleavage and nuclear translocation in disease states. Both CRMP-2 (Zhang et al, 2007) and CRMP-3 (Hou et al, 2006) are cleaved by calpain in vitro after excitotoxic injury and in vivo after traumatic or ischemic brain injury, respectively. Calpain cleavage of both CRMP-2 and CRMP-3 leads to their translocation from the cytosol to the nucleus. Furthermore, overexpression of the calpain-cleaved form of CRMP-3 was sufficient to induce neuronal death, whereas knockdown of endogenous CRMP-3 was neuroprotective (Hou et al, 2006), suggesting a causal role of nuclear CRMP in neurodegeneration.
Summary
This review has attempted to outline the potential mechanisms by which pathologic calpain activity can contribute to postischemic neurodegeneration. Key considerations include the potential role of specific calpain isoforms, subcellular localization, and the dysregulatory effect that calpain-mediated proteolysis has on a wide variety of substrates involved in both essential cell functions and cell death signaling. The sensitivity of most calcium regulatory proteins to calpain-mediated proteolysis is the foundation for hypothesized feed-forward mechanisms by which pathologic calpain activity causes lethal neuronal calcium overload (Table 2). However, direct in vivo evidence is still lacking. Furthermore, disruption of neuronal calcium homeostasis is only one mechanism by which calpains have been proposed to cause cell death. Disruption of survival signals or activation of death signals (Table 3) through cytosolic, mitochondrial, lysosomal, and nuclear substrates remains as potential mechanisms that require further evaluation (Figure 2).
Moving forward, the field is likely to benefit from more effective and specific strategies to inhibit postischemic activity of various calpain isoforms in vivo. Furthermore, elucidation of the discrete pathways that contribute to neurodegeneration will require much more comprehensive and precise evaluation of the calpain substrates that are actually cleaved in postischemic neurons. Perhaps the greatest challenge is to elucidate the relative contribution of each cleaved substrate to subsequent neuronal death. Finally, the physiologic function of the calpain—calpastatin system cannot be ignored. Given the known role of calpains in key cell processes such as mitosis, nonspecific calpain inhibition is a potentially unacceptable therapeutic strategy. Approaches that might avoid toxicity include isoform-specific calpain inhibition, targeting nondividing cells, or targeting subcellular compartments where pathologic calpain activity contributes to neuronal death.