
Abstract
The molecular biological techniques for plasmid-based assembly and cloning of gene open reading frames are essential for elucidating the function of the proteins encoded by the genes. High-throughput integrated robotic molecular biology platforms that have the capacity to rapidly clone and express heterologous gene open reading frames in bacteria and yeast and to screen large numbers of expressed proteins for optimized function are an important technology for improving microbial strains Published by Elsevier Inc. on behalf of the Society for Laboratory Automation and Screening for biofuel production. The process involves the production of full-length complementary DNA libraries as a source of plasmid-based clones to express the desired proteins in active form for determination of their functions. Proteins that were identified by high-throughput screening as having desired characteristics are overexpressed in microbes to enable them to perform functions that will allow more cost-effective and sustainable production of biofuels. Because the plasmid libraries are composed of several thousand unique genes, automation of the process is essential. This review describes the design and implementation of an automated integrated programmable robotic workcell capable of producing complementary DNA libraries, colony picking, isolating plasmid DNA, transforming yeast and bacteria, expressing protein, and performing appropriate functional assays. These operations will allow tailoring microbial strains to use renewable feedstocks for production of biofuels, bioderived chemicals, fertilizers, and other coproducts for profitable and sustainable biorefineries.
Keywords
Introduction
High-throughput platforms that can rapidly clone and express heterologous gene open reading frames (ORFs) in bacteria and yeast and can screen large numbers of proteins expressed by these genes for optimized function are important for improving microbial strains for bioenergy applications. Combined with rapid gene assembly and mutagenesis strategies, gene ORFs can be synthesized, cloned into plas-mid vectors, transformed into yeast strains, and screened to identify those that express proteins that give increased fuel ethanol production, allow coproduction of biodiesel, enable use of biomass as a feedstock, and express valuable bioderived coproducts.
The molecular biological techniques for plasmid-based assembly and cloning of gene ORFs are essential for elucidating the function of the proteins encoded by the genes. These techniques involve the production of full-length complementary DNA (cDNA) libraries as a source of plasmid-based clones to express the desired proteins in active form for determination of their functions.1–8 This field of study, known as plasmid-based functional proteomics, requires rapid plasmid preparation methods to obtain adequate quantities of high-quality plasmid DNA to conduct all required steps in the process from creation of plasmid libraries to functional testing of expressed proteins.
Because the plasmid libraries are composed of several thousand unique genes, automation of the process is essential. 9–21 The ideal system would be an automated integrated programmable platform capable of producing full-length cDNA libraries, colony picking, isolating plasmid DNA, transforming yeast and bacteria, expressing protein, and performing appropriate functional assays. Such an automated system requires the integration of different types of equipment and instruments with the desired capabilities.
An example of this system is the integrated plasmid-based functional proteomic robotic workcell built at United States Department of Agriculture, Agricultural Research Service, National Center for Agricultural Utilization Research (USDA, ARS, NCAUR) in Peoria, Illinois. The workcell automates all the required tasks from plasmid library creation through functional testing of the expressed protein(s) for large sets of clones. The robotic workcell (Fig. 1) was designed and built by USDA and Hudson Robotics, Inc. (formerly Hudson Control Group, Inc.) to conduct plasmid-based functional proteomics for optimization of gene ORFs encoding proteins of interest for advanced bioenergy applications.
Schematic and picture of integrated robotic workcell. The position numbers are identified as follows: # IA: Track I; # I B: Track 2; #2A StackLink (Track I); #2B: StackLink (Track 2); #3: 4-Axis PlateCrane EX; #4: Colony picker/arrayer; #5: PCR thermal cycler with autolid; #6: UV/VIS Plate Reader; #7: ABgenePlate sealer (foil); #8: Plate sealer (porous tape); #9: Liquid handler with centrifuge; # 10: Hudson micro 10 filler; # 11: Hudson plate aspirator; # 12: Automated incubator; # 13: Passive stackers; # 14: Computer and monitor; # 15: Barcode reader; #SI to #S6: StopLink plate positions, StopLink plate. Reprinted from Proteome Science, vol 4, Hughes, S. R.; Riedmuller, S. B.; Mertens, J. A.; Li, X.-L; Bischoff, K.M; Qureshi, N.; Cotta, M. A; Farrelly, P. J., High-throughput screening of cellulase F mutants from multiplexed plasmid sets using an automated plate assay on a functional proteomic robotic workcell, pages 1-14, Copyright 2006, with permission from BioMed Central.
Components of an Integrated Plasmid-Based Robotic Workcell
Liquid Handler for Plasmid Preparation
The initial phase of the project was designing and assembling a track-based liquid handler for 96-well format plasmid preparation. The liquid handler was incorporated into the robotic workcell, and it was used to provide plasmid to test the molecular biology protocols for the workcell. The design allowed processing of up to four 96-well plates in one run. To evaluate liquid handler operation, mutagenized cellulase F (CelF) ORFs were generated by site-directed polymerase chain reaction (PCR) mutagenesis from the wild-type CelF ORF, 22 an endoglucanase enzyme from the anaerobic fungus Orpinomyces PC-2, to obtain clones expressing optimized cellulases, and a plasmid library of these mutagenized ORFs was prepared using a novel Invitrogen Gateway cloning strategy. 23 Cellulases are capable of digesting plant cell wall polysaccharides and therefore have potential industrial application in breaking down plant biomass for use in fuel ethanol production.
The quantity of plasmid produced using the robotic liquid handler allowed several parameters to be measured on one set of samples. A small starting culture was used (1.347 mL), and the resulting bacterial lysate containing the plasmid was processed. An average volume of 160 μL of plasmid was collected. The average amount of plasmid obtained was 5.35 μg per well for the automated process. No signs of contamination were detected between wells processed on the robotic workcell. The entire four × 96-well plate process on the liquid handler, including long- and short-term storage, pelleting of the incoming bacterial cultures, execution of all steps in a sterile fashion with reusable pipette tips, integrated into the robotic workcell, can deliver plates to the workcell in about 6 h. Automated plasmid production by the liquid handler was shown to be robust and repeatable, and plasmid quality was equivalent to that of manual operation. The plasmids were used in various expression strategies to produce protein for functional assays to assess protein activity and level of expression in vitro and in vivo. On an average, 88 μL of the plasmid set remained and could be stored for future reactions. A liquid handler of this speed and capability is able to perform all protocols necessary for the plasmid-based functional proteomic robotic workcell. The quality and quantity of plasmids prepared on the liquid handler enabled implementation of subsequent workcell protocols, including DNA sequencing, in vitro transcription/translation, and transformation of bacterial and yeast strains for protein expression. 23
Robotic Colony Picker for Multiplex Screening Format
Producing recombinant proteins from large-scale plasmid preparations using automated molecular biology techniques for high-throughput screening requires expensive reagents. A multiplex format can reduce the cost of the process by decreasing the number of wells to be screened and thus decreasing the amount of materials needed. An automated multiplex method was developed by integration of a robotic colony-picking component onto the automated workcell platform for high-throughput screening of a plasmid library of clones expressing mutants of CelF to obtain clones expressing optimized cellulases. 24 The multiplex method involves an initial screening of a multiplexed culture that contains a mixture of clones. Individual clones giving rise to improved cellulase mutants are then isolated by picking single colonies from the multiplexed cultures, producing plasmid DNA from the picked colonies, and expressing protein in automated in vitro transcription/translation reactions to identify mutants with improved activity versus wild-type CelF on a plate-based functional assay.
A plasmid library of mutagenized clones of the celF gene with targeted variations in the last four codons, constructed by site-directed PCR mutagenesis, was transformed into Escherichia coli. The robotic colony picker integrated into the workcell was used to inoculate medium in a 960-well deep-well plate, combining eight transformants per well into a multiplexed set, and the plate was incubated on the work-cell. Using the liquid handler component of the workcell, plasmids were prepared from the multiplexed culture and used for in vitro transcription and translation. The multiplexed expressed recombinant proteins were screened versus the wild type for improved activity and stability in an azo-carboxymethyl cellulose (CMC) plate assay. Five multiplexed cultures were identified as containing mutants having improved activity. Control experiments were performed to determine the effect of increasing amounts of CelF mutants in a multiplexed well or increasing numbers of multiplexed colonies on assay sensitivity. It was demonstrated that if the number of colonies multiplexed in a well is increased relative to an individual CelF mutant, the activity of that mutant is still detectable. In addition, it was also shown that as the amount of an individual active CelF mutant spotted on the azo-CMC (Megazyme International Ireland Ltd., Bray, Ireland) plate is increased up to 10-fold, the activity measured by the diameter of the cleared zone produced on the plate increases in a linear fashion. 24
The multiplexed wells containing mutants with high activity that were identified on the multiplexed azo-CMC screening plate were linked back to the corresponding multiplexed cultures stored in glycerol. Individual clones were then isolated from the multiplexed cultures using the workcell to inoculate single cultures from stock spread plates, prepare plasmid, produce recombinant protein, and assay for activity, performing all operations on an integrated automated platform. The screening assay and subsequent isolation and assay of individual clones resulted in identification of four CelF mutants with higher activity and greater thermal and pH stability than wild-type CelF, demonstrating that the multiplex method using an integrated automated workcell for high-throughput screening in a functional proteomic assay increases the numbers of clones that can be screened and permits rapid identification of optimized clones.
Automated Molecular Biology Protocols for All Operations
Automation is essential for the production and screening of large numbers of expression-ready plasmid sets used to develop optimized clones and improved microbial strains. An important application of such an automated platform is in the development and screening of optimized genes in high throughput for use in the production of improved commercial yeast strains to convert biomass to ethanol. These strains are being engineered to express genes for hydrolysis and fermentation of cellulose or hemicellulose to ethanol. At the same time, these strains can provide host capability for expression of high-value proteins and peptides, such as a bioinsecticide. Genes for these proteins can be mutagenized and screened in high throughput to optimize the desired functional characteristics. A set of automated molecular biology protocols, including assembly of mutagenized gene sequences, purification of PCR amplicons, ligation of PCR products into vectors, transformation of competent E. coli, plating of recovered transformants, and inoculation of cultures for plasmid preparation, was developed for the plasmid-based integrated robotic workcell.23,24,26
Programmable Software for Integration of All Operations
The molecular biology protocols were scripted for the robotic workcell using TrackLink and SoftLinx software developed by Hudson Robotics, Inc. (Springfield, NJ) to integrate all plasmid-based operations on the workcell. The following is a brief outline of the operations from preparation of plasmids from cells transformed with vectors containing the gene ORF of interest to expression of protein for evaluation. A clean 96-well plate is programmed to move along a track on the workcell from the stacker area to the filler position for filling with medium, and then it is moved to the liquid handler deck by the Plate Crane robotic arm for inoculation using the liquid handler. The plate is moved along a track to the colony picker for preparation of spread plates. The spread plate is moved to the sealer, and after sealing, it is placed in the incubator. Four clean 24-well deep-well plates are moved by track from the stacker to the filler for addition of medium and then moved by the plate crane to the deck for inoculation from the cultures on the 96-well plate. The deep-well plates are moved by track to the incubator. After incubation, the plates are moved to the liquid handler for plasmid preparation, and the plasmids are eluted into a collection plate. Plasmid concentrations are analyzed on the workcell by adding samples from the collection plate to a ultraviolet (UV) optically clear 96-well microplate, and the readings are taken at A230, A260, and A280 in the UV/visible (VIS) microplate reader (Fig. 1 #6). Additional spectrographic analysis of plasmid samples was performed using a fiber optic spectrophotometer to develop a factor to adjust for variances in the height of the readings taken by the UV/VIS microplate reader. 24 Protein expression reactions are prepared in the plasmid plates on the deck of the liquid handler, mixed by aspiration, then moved by track into position for transport by the plate crane to the heat sealer and finally into the thermal cycler for incubation.
A variety of plasmids can be prepared using the same programmable software. The gene ORFs of interest are first used in a topoisomerase (TOPO) ligation with the Gateway-adapted plasmid pENTR D TOPO (Invitrogen Corporation, Carlsbad, CA) for directional cloning and transformed into TOP 10 E. coli cells. After the pENTR D TOPO plasmids containing the gene ORF of interest are collected, LR clonase reactions can be performed to move the gene ORF insert into either pEXP-1 DEST for in vitro expression in bacterial lysate containing T7 polymerase or into pDEST 17 for in vivo expression in bacteria or pYES2 DEST 52 for in vivo expression in yeast. 23 The gene ORFs can also be cloned into Gateway-adapted vectors, such as the yeast expression vector portable small ubiquitin-like modifier (pSUMO) duo that can be used for in vitro and in vivo bacterial and in vivo yeast expression. 26 Expressed proteins are spotted by the pipette arm of the liquid handler on assay plates moved into position on the liquid handler deck, and the proteins are then assayed for function. In addition to the CMC (agar) plate assay coupled with a digital imaging station described here, the robotic workcell could be programmed to perform enzyme assays in solution and collect data with the UV/VIS Plate Reader (BioTek Instruments, Inc., Winooski, VT).
Applications of the Integrated Plasmid-Based Robotic Workcell
Protocols for Plasmid Library Preparation
To demonstrate the application of an automated molecular biology protocol, a plasmid library of gene ORFs encoding mutants of a bioinsecticide, lycotoxin-1 (Lyt-1), from wolf spider (Lycosa carolinensis), which is highly effective against insects but not toxic to humans 25 was produced in the pENTR D TOPO vector using PCR mutagenesis in an amino acid scanning strategy to generate a complete set of mutations across the Lyt-1 gene. The protocols were used on the integrated plasmid-based robotic workcell to assemble and purify the mutagenized inserts, ligate these inserts at high efficiency into a TOPO cloning vector, transform these libraries in high throughput into E. coli, inoculate plates for plasmid preparation, and recover the plasmids all in a fully automated fashion. A variation of the multiplex method 24 that was made possible by integration of a robotic colony-picking component onto an automated workcell platform was used. 26 These protocols form the core of a fully automated molecular biology platform. Fully automated molecular biology protocols are essential to allow rapid production of PCR-generated inserts for libraries, whether cDNA libraries or libraries of mutagenized clones, for incorporation into vectors and ultimately plasmid recovery. A protocol for amino acid scanning mutagenesis (AASM) was used to generate the complete set of mutations across the Lyt-1 gene library. 27 The resulting pENTR D TOPO libraries of PCR-assembled or AASM-mutagenized products can be recombi-nationally cloned into Gateway-adapted vectors, such as the yeast expression vector pSUMO duo that can be used for in vitro and in vivo bacterial and in vivo yeast expression. 28
AASM Protocol
The first step in the AASM protocol is an assembly strategy using overlapping oligonucleotides of 50 bp in length to assemble the clone for the gene of interest. A second set of oligonucleotides is produced to assemble a clone with an identical sequence, but the overlap is offset by 25 bp. Once these two clone sets are produced, there is no section of the clone that is not covered by an overlap when introduction of an NNN—NNN—NNN—NNN set of randomization co-dons is shifted down along both clone set sequences leaving at least 10-bp overlap at the 3’ end of each oligonucleotide. This NNN—NNN—NNN—NNN codon randomization set in one oligonucleotide substituted into each of the two identical assembled clones will give rise to 204 = 160,000 variants for all 20 amino acids at each of the positions in the expressed mutant protein corresponding to these four codons. This four-codon substitution yields a manageable number of clones to screen for the average gene length of 1000 bp (also for smaller and larger genes).
An example of AASM strategy using the Lyt-1 gene sequence that codes for a 25-amino acid protein is shown in Figure 2. Each codon in the Lyt-1 sequence is replaced by NNN, where N is any of the possible nucleotides. Each synthetic gene is produced from a 55-nucleotide forward oligonucleotide annealed to an 87-nucleotide reverse oligonucleotide with a 30-bp overlap. The forward oligonucleotide contains the codons for the TOPO directional cloning signal, the 6X HIS tag, all four codons of the enterokinase K (Ent K) site, and seven mutagenized codons of the Lyt-1 sequence. The 25 mutagenized reverse oligonucleotides (overlapping by 30 bp) contain three codons of the Ent K site and the NNN substitutions in the codons for each amino acid in the Lyt-1 protein.
26
The level of screening for the mutants produced by this strategy can be conducted on most liquid handler-based proteomic workcell robotic platforms on the market, including the unit at NCAUR.
AASM strategy using the Lyt-1 gene sequence that codes for a 25-amino acid protein. Each codon in the Lyt-1 sequence is replaced by NNN, where N is any of the possible nucleotides. Each synthetic gene is produced from a 55-nucleotide forward oligonucleotide annealed to an 87-nucleotide reverse oligonucleotide with a 30-bp overlap. The forward oligonucleotide contains the codons for the TOPO directional cloning signal, the 6X HIS tag, all four codons of the Ent K site, and seven mutagenized codons of the Lyt-1 sequence. The 25 mutagenized reverse oligonucleotides (overlapping by 30 bp) contain three codons of the Ent K site and the NNN substitutions in the codons for each amino acid in the Lyt-1 protein. Reprinted from Journal of the Association for Laboratory Automation, vol 12, Hughes, S. R.; Dowd, P. F.; Hector, R. E.; Riedmuller, S. B.; Bartolett, S.; Mertens, J. A.; Qureshi, N.; Liu, S.; Bischoff, K. M.; Li, X.-L; Jackson, J. S. Jr.; Sterner, D.; Panavas, T.; Rich, J. O.; Farrelly, P. J.; Butt, T. R.; Cotta, M. A., Cost-effective high-throughput fully automated construction of a multiplex library of mutagenized open reading frames for an insecticidal peptide using a plasmid-based functional proteomic robotic workcell with improved vacuum system, pages 202-212, Copyright 2007.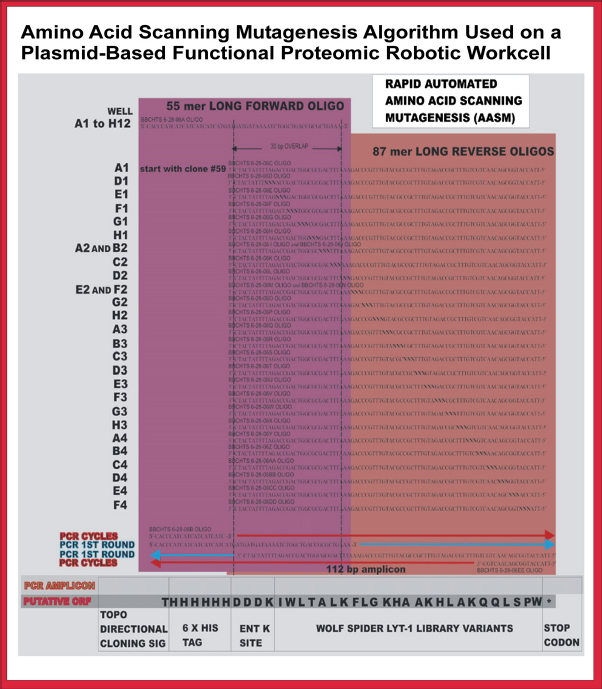
The library of mutagenized genes is expressed using additional automated molecular biology routines either in vivo or in vitro on the integrated robotic platform, and the expressed peptides or proteins are screened using various assays, either with intact cells expressing the mutant gene products or with mutants produced by in vitro expression to identify mutants with optimal characteristics. After optimized clones for these mutants are identified, they are selected and used in a combinatorial algorithm to evaluate all possible combinations of test mutations in the applicable assay.
The AASM process is much faster and cheaper than randomized mutagenesis because the changes are produced in a systematic fashion along the entire sequence of the clone, and mutations are forced to occur at sites that may not change during random mutagenesis. It also has advantages over targeted mutagenesis because the randomized sites in AASM are screened with a functional proteomic assay that selects the randomized oligonucleotides that produce an optimized clone and indicates on which regions to focus. It is also possible to combine the various improved randomized oligonucleotides from AASM to find those combinations that might have a particular synergy to generate a superior optimized ORF.
Protein Expression and Screening Operations Using Integrated Track-Based Programs
An example of an integrated operation scripted for the plasmid-based robotic workcell was the functional proteomic assay in a multiplexed setting for high-throughput screening of mutants of CelF to identify plasmids containing optimized clones expressing mutants with improved activity at lower pH. 24 The automated track-based operations, which are described in detail in the following sections, included picking of colonies into a 96-well plate, inoculation from the 96-well plate into four 24-well deep-well plates in quadruplicate, in vitro protein production, and evaluation of the expressed protein for temperature and pH stability using an azo-CMC plate assay.
Automated Picking of Colonies Into 96-Well Plate.
The spread plates containing the colonies from the multiplex cultures were placed onto the workcell in the passive stacker (Fig. 1 #13). An ABgene 96-well deep-well plate (Thermo Fisher Scientific, Rockford, IL) was loaded into the active stacker, a 4-L carboy containing 2 L of medium was connected to the Hudson micro l0 filler (Fig. 1 #10), and the automated protocol for picking colonies into the 96-well plate was initiated. The ABgene 96-well deep-well plate was sent from the Track 2 active stacker (Fig. 1 #2B) to the Hudson micro 10 filler (position S3). Each well was filled with 1.6 mL of medium, and the plate moved to the Track 2 Plate Crane StopLink position S2. The plate was moved from position S2 to S1 by the plate crane (Fig. 1 #3) and then sent to S6 for inoculation with each of the four control cultures from the cold tube rack on the liquid handler deck (Fig. 1 #9) into the first six wells in the two upper left hand rows in blocks of two or four using the Hudson liquid handler (Xantus Liquid Handler; Sias AG, Hombrechtikon, Switzerland, adapted by Hudson Robotics, Inc.). After the controls were added, the plate was sent via a Hudson TrackLink (Fig. 1 #1A) into the deck space of the picker in StopLink position S4. From this location, the gripper tool of the picker (Fig. 1 #4) moved the deep-well plate containing medium into the picker destination location and removed the lid to receive colonies. Picking was performed using a Hudson SoftLinx adapted routine of the BioRad VersArray picker integrated into the workcell. The spread plates for the identified wells were moved from the Hudson passive stacker (Fig. 1 #13) by the plate crane (Fig. 1 #3) to S1. SoftLinx software from Hudson was used to identify spread plate wells as the plates were moved onto the deck of the picker passing the bar code reader on Track 1 (Fig. 1 #1A and #15). The plate was then moved to S4 where the gripper tool on the picker took the colony plate into the lighted area of the picker deck (Fig. 1 #4). The charge-coupled device camera photographed the spread plate in the lighted area to record a digital image for capture and recognition of stereotactic coordinates for all colonies of the programmed size range. The colonies were picked using four dedicated pins of the 16-pin picking head (Fig. 1 #4) and inoculated, four colonies at a time, in an S-shaped pattern into the 96-well plate, randomly taking 20 colonies from each of the first four spread plates and four colonies from the last spread plate. The inoculated plate was taken from the destination location to S1, picked up, placed into the Brandel RS3000 (Brandel, Gaithersburg, MD) porous tape sealer (Fig. 1 #8) and sealed with gas-permeable tape. The plate was moved to S2 and loaded into the Liconic incubator (Fig. 1 #12) at the modified StopLink S5 and incubated for 30 h at 37 °C and 600 rpm.
Automated Inoculation From 96-Well Plate Into Four 24-Well Plates in Quadruplicate (384 Wells).
This protocol was carried out with a modified SoftLinx integration routine (Fig. 1 #14). Qiagen 24-well deep-well plates were loaded into the active stacker (Fig. 1 #2B), a 4-L sterile carboy containing 2 L of medium was attached to the filler (Fig. 1 #10) with sterile tubing, and the automated protocol for liquid culture inoculation into 24-well plates was initiated. The 24-well deep-well plates were sent from the Track 2 active stacker (Fig. 1 #2B) to the micro 10 filler (position S3), and each well was filled with 5 mL of medium. The plates were moved from S3 to Track 2 position S2 then to Track 1 position S1 using the PlateCrane EX (Fig. 1 #3) and sent via a Hudson TrackLink to position S6 and then onto the deck of the liquid handler unit (Fig. 1 #9) of the workcell using the gripper arm of the liquid handler. Four 24-well plates at a time were inoculated with 20-μL culture in each well in the image of the 96-well plate four times to give quadruplicate sets of four 24-well plates representing the controls and the 84 colonies picked. The inoculated plates were taken to position S6 then moved to S1 on Track 1, picked up by the plate crane, placed into the porous tape sealer (Brandel RS3000) (Fig. 1 #8) on the workcell, and each plate was sealed with gas-permeable tape. All the plates were moved to position S2 on Track 2 and then to S6 and into the Liconic incubator for 30 h at 37 °C and 600 rpm. Large-scale plasmid preparation was then carried out on the liquid handler as described previously. 23 These plasmids were eluted on the workcell into one Matrix 2D bar-coded collection plate, samples were placed in a UV/VIS microplate (Fig. 1 #6), and the concentrations were adjusted for consistent expression of the proteins from the plasmids. 24
Automated In Vitro Protein Production.
Protein production was initiated by placing Hard-Shell 96-well skirted low-profile PCR plates (Bio-Rad Life Science, Hercules, CA) and UV/VIS optically clear standard microplates (Thermo Fisher Scientific, Rockford, IL) into the active stacker (Fig. 1 #2A). Protein reagent was filled into Matrix 2D bar-coded tubes (Thermo Fisher Scientific) and placed into the cold reagent position on the liquid handler (Fig. 1 #9) for holding at 2 °C. The protocol for protein production adapted from SoftLinx, accessing Xanthus Application Protocol routines for reaction set up and spotting (Fig. 1 #14), was initiated. Protein reactions were prepared from plates of plasmid on the deck of the liquid handler, then a new Hard-Shell plate was moved from the active stacker to S6 on the liquid handler deck and transported by the liquid handler gripper to the cold block for reagent additions. The reagents were mixed by aspiration, and the plate was moved to S6 then to position S1 for transport by the plate crane into the ABgene 300 foil heat sealer (Fig. 1 #7) and finally into the PCR thermal cycler (Fig. 1 #5). After the plasmids were obtained using the automated plasmid preparation protocol for large-scale plasmid preparation, 5-μL aliquots of the mutagenized library plasmids was used for in vitro transcription/translation reactions to generate recombinant protein. Reactions were incubated for 10 h at 30 °C in the PCR thermal cycler after sealing with foil tape. The temperature was then held at 4 °C until the azo-CMC plate assay for activity of the proteins produced could be initiated. 24
Temperature and pH Evaluations of Expressed Protein Using Automated azo-CMC Plate Assay.
For pH evaluation of the mutant proteins, four azo-CMC plates at pH 5.8, 5.0, 4.5, or 4.0 were loaded into the passive stacker (Fig. 1 #13), delidded by the gripper arm of the plate crane (Fig. 1 #3) and moved to S1, then to S6 into the working area of the liquid handler (Fig. 1 #9) for the automated azo-CMC plate assay. The plate crane moved the 96-well hardshell plate containing the in vitro-produced protein from the PCR thermal cycler (Fig. 1 #5) to S1. From this position, the plate was taken to S6 so that the liquid handler gripper could place it into the cold plate position on the deck, then into the jig position for piercing and finally back to the cold position on the deck. A SoftLinx-initiated Xanthus Application Protocol was used to spot protein on the azo-CMC plates for assay using the pipette arm of the liquid handler. Plates were moved to S6 with the gripper, then sent to S1 to be picked up by the plate crane and placed on S2. The plates were moved to S5 for loading into the incubator. For evaluation of temperature stability, the hardshell plate with protein was moved to the PCR thermal cycler (Fig. 1 #5), incubated at 50 °C for 1 h then moved back to the cold position on the liquid handler deck. A new pH 5.8 azo-CMC plate was moved into the working area, spotted with the heated proteins, and moved to the Liconic incubator with the other azo-CMC plates. All the plates were incubated at 37 °C for 10 h. Plates were moved to the out stack on the passive stacker and photographed using the Alpha Innotech 3400 digital imaging station. Activity of the protein mutants was determined by their reaction with azo-CMC on the plates. 24
Genetic Engineering of Yeast Using Selected Genes for Xylose Utilization
Fuel ethanol production from biomass at the industrial level using Saccharomyces cerevisiae shows great promise for satisfying future energy demands, but the limited range of materials that can be fermented remains an obstacle to cost-effective bioethanol production.29,30 Although several genetically engineered strains of S. cerevisiae have been developed that will ferment xylose to ethanol,31–34 further optimization is needed. It will require the simultaneous expression, at sufficiently high level, of all the enzymes and proteins needed to allow industrial yeast strains to grow efficiently on pentose and hexose sugars anaerobically. In addition, for cost-effective industrial ethanol production from biomass, it will be necessary to express the enzymes required to hydrolyze the lignocellulosic feedstocks that are the source of hexose and pentose sugars. Genes considered necessary for complete fermentation of xylose and arabinose, the two major pentose sugar constituents of lignocellulosic biomass, include those encoding xylose isomerase (XI), xylulokinase (XKS), arabinose A, arabinose B, and arabinose D,33,34 which may be obtained from a microorganism naturally capable of fermenting these sugars. Obtaining the sugar constituents of lignocellulosic feedstocks also requires utilization of hydrolytic enzymes, including cellu-ases and hemicellulases after initial chemical pretreat-ment.35,36 The cost-effectiveness of the ethanol fermentation process could also be enhanced by obtaining high-value co-products and byproducts from the process, such as monomers for polymer production and commercially important proteins and peptides. Genes for these proteins and peptides can be mu-tagenized, placed in an expression system capable of producing high levels of functional proteins or peptides, and screened in high throughput to optimize desired characteristics.
A three-plasmid yeast expression system using the pSUMO vector set combined with the efficient endogenous yeast protease U1p1 was developed28,37-39 for production of large amounts of soluble functional protein in S. cerevisiae.40,41 Each vector has a different selectable marker (URA, TRP, or LEU), and the system provides high expression levels of three different proteins simultaneously. Expression levels of the His-tagged proteins were determined from Western blots of the proteins run on 16% polyacrylamide gel using AlphaEaseFC software for Windows 2000 to determine band density compared with known molecular weight markers from the Qiagen 6xHis-tagged set treated in the same fashion as the XI and Lyt-1 protein samples. Protein concentrations for the individual XI and Lyt-1 bands for the two-plasmid strain are on the order of 12—14 μg/mL. For the three-plasmid strain, expression of Lyt-1 is slightly greater than that of XI, with the Lyt-1 concentrations ranging from 14 to 19 μg/mL and the XI concentrations from 9 to 12 μg/mL. However, the sum of the concentrations determined for the XI and Lyt-1 bands is similar in all strains expressing them, about 25—29 μg/mL. The concentrations of XKS and transal-dolase (TAL) cannot be determined directly because they are not His-tagged, but the increased concentration of the His-tagged SUMO band (14—16 μg/mL) in the triply transformed yeast strains over that in the doubly transformed yeast strains (8—9 μg/mL) gives a measure of the expression of XKS or TAL.
42
This system was integrated into the protocols on an automated plasmid-based robotic platform to screen engineered strains of S. cerevisiae for improved growth on xylose (Fig. 3).
42
A three-plasmid yeast expression system using the pSUMO vector set for high expression levels of three different proteins integrated into the protocols on an automated plasmid-based robotic platform to screen engineered S. cerevisiae strains for improved growth on xylose. Step I: Assembly of His-tagged XI ORF and cloning into pSUMOduo/URA (vector I). Step 2: Gene optimization using AASM to randomize Lyt-1 at each of 25 positions for all 20 possible amino acids in the HisEntKLyt-1 fusion peptide and cloning into pSUMOduo/TRP (vector 2). Step 3: Cloning of additional genes important for xylose utilization into pSUMOduo/LEU (vector 3). Reprinted from Plasmid, vol 61, Hughes, S. R.; Sterner, D. E.; Bischoff, K. M.; Hector, R. E.; Dowd, P. F.; Qureshi, N.; Bang, S. S.; Grynaviski, N.; Chakrabarty, T.; Johnson, E. T.; Dien, B. S.; Mertens, J. A.; Caughey, R. J.; Liu, S.; Butt, T. R.; LaBaer, J.; Cotta, M. A.; Rich, J. O., Engineered Saccharomyces cerevisiae strain for improved xylose utilization with a three-plasmid SUMO yeast expression system, pages 22-38, Copyright 2008.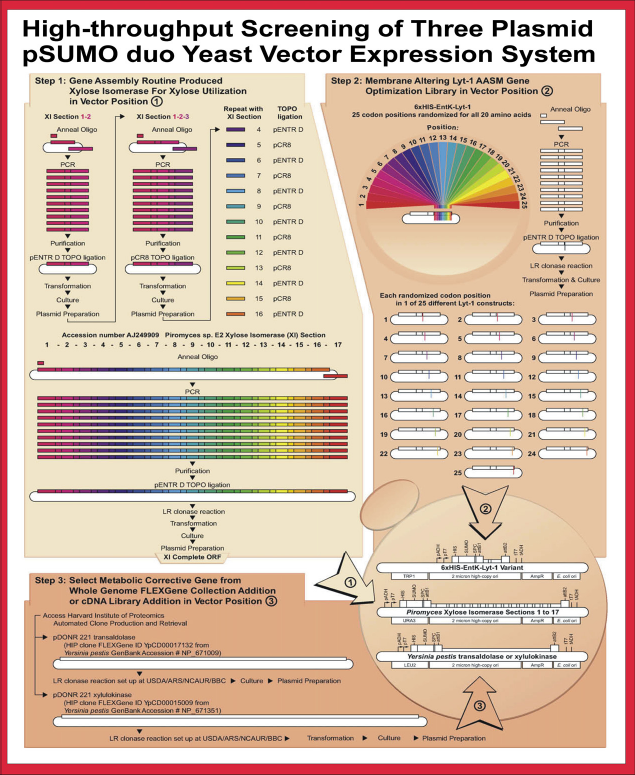
First, a novel PCR assembly strategy was used to clone a Piromyces sp. E2 XI gene ORF into the URA-selectable SUMO vector and the plasmid was placed into the S. cerevisiae INVScl strain, 23 a fast-growing diploid strain ideal for expression,43–45 to give the strain designated INVSc1-XI. Second, AASM was used to generate a library of mutagenized genes,26,27 encoding the bioinsecticidal peptide Lyt-1, and the library was cloned into the TRP-selectable SUMO vector and placed into INVScl-XI to give the strain designated INVSc1-XI-Lyt-1. Third, the XKS gene of Yersinia pestis was moved from a pDONR221 collection 46 and cloned into the LEU-selectable SUMO vector and placed into the INVScl-XI-Lyt-1 yeast. Yeast strains expressing XI and XKS with or without Lyt-1 showed improved growth on xylose compared with INVScl-XI yeast. The vectors contain the high-copy 2-μm origin of replication to give a copy number of roughly 20 per yeast cell. 47 Expression of XI and XKS is suggested as a means of enabling yeast to metabolize xylose more rapidly through the pentose phosphate pathway.44,45,48—54
The SUMO plasmids are particularly well suited for integration with the automated protocols on the robotic platform and complement the PCR assembly and TOPO directional in-frame cloning strategy. 23 This set of plasmids used on the automated platform23,24,26 offers the possibility of expressing pentose-utilization enzymes and commercially important peptides in yeast or to introduce other enzymes, such as cellulases, 55 to produce improved yeast strains for industrial use and screening the resulting yeast strains in high throughput for those that grow rapidly anaerobically and produce ethanol at sufficiently high levels for industrial application.
Genetic Engineering of Yeast Using Full-Genome Collection of Yeast Gene ORFs to Improve Xylose Utilization
Numerous attempts have been made to engineer microorganisms capable of efficient fermentation of both glucose and xylose as a means of achieving economically feasible biomass conversion into fuel ethanol. These attempts have focused on engineering the glucose-fermenting industrial yeast S. cerevisiae to use pentose sugars from lignocellulosic biomass by introducing the enzymes of the initial stages of xylose metabolism.32,51 One approach has been to engineer S. cerevisiae to express XI, which catalyzes the conversion of xylose to xylulose and does not require redox cofactors. Introduction of a functional XI into S. cerevisiae allows slow metabolism of xylose but is not sufficient for high rates of anaerobic xylose fermentation.44,45,51,54 It is significant that engineering approaches to improve pentose-fermenting yeasts have required expression of auxiliary genes to complement activity of XL 48 Despite evidence that overexpression of further genes is required, no systematic screening of the yeast genome has been undertaken to identify the genes that need to be overexpressed for improved xylose fermentation.
A study was performed to evaluate overexpression of each S. cerevisiae gene ORF in a strain also expressing XI and determine which of the genes, if any, confer the ability for anaerobic growth on xylose.
56
These genes would be appropriate targets for further improving the fermentation characteristics of xylose-fermenting Saccharomyces strains. The study used a collection of S. cerevisiae gene ORFs in pOAD LEU-selectable vectors driven by an alcohol dehydrogenase (ADH) promoter in the PJ69-4 MATa S. cerevisiae strain.57,58 Each of these was mated to the haploid PJ69-4 MATalpha S. cerevisiae strain containing the Piromyces sp. E2 XI gene
27
expressed from a pDEST32 TRP-selectable vector with an ADH promoter.
56
To mate and screen the entire collection of gene ORFs, an automated high-throughput strategy incorporating the essential features of the conventional manual process was developed and implemented on an integrated robotic workcell (Fig. 4).
Steps required for mating the PJ69-4 MATalpha haploid yeast strain, expressing the Piromyces XI gene from the plasmid pDEST32, to the PJ69-4 MATa haploid yeast strain, expressing one of the collection of yeast genes from the plasmid pOAD. The pDEST32-XI TRP-selectable plasmid was constructed by replacing the LEU2 gene in the pDEST32 LEU2-selectable bait plasmid commercially available in the ProQuest Two-Hybrid System kit with the TRPI gene. The PiromycesXI gene was cloned into the resulting TRP-selectable modified bait plasmid using standard Gateway recombination procedures with LR clonase. A collection of PCR-generated ORFs predicted from the S. cerevisiae genome in pOAD LEU-selectable vectors transformed into PJ69-4 MATa haploid yeast strain was provided by Dr. Stanley Fields at University of Washington, Seattle, Washington. Mating of PJ69-4 MATa cells containing pOAD-gene ORF LEU-selectable plasmids with MATalpha PJ69-4 yeast strain containing pDEST32-XI TRP-selectable plasmids was performed on the liquid handler. Double-plasmid strains were derived from mating PJ69-4 MATalpha strain expressing Piromyces XI with PJ69-4 MATa strain expressing one of the yeast genes. Triple-plasmid strains were produced by transformation of double-plasmid strains with pSUMOduo-RGStetHisXKS URA-selectable plasmid. Reprinted from Journal of the Association for Laboratory Automation, vol 14, Hughes, S. R.; Rich, J. O.; Bischoff, K. M.; Hector, R. E.; Qureshi, N.; Saha, B. C.; Dien, B. S.; Liu S.; Jackson, J. S.; Sterner, D. E.; Butt, T. R.; LaBaer, J.; Cotta, M. A., Automated yeast transformation protocol to engineer Saccharomyces cerevisiae strains for improved cellulosic ethanol production with open reading frames that express proteins binding to xylose isomerase identified using a robotic two-hybrid screen, pages 200-212, Copyright 2009.
The resulting 6113 mated diploid strains containing the XI gene and a different yeast gene ORF were screened for growth on xylose in anaerobic plate cultures using the integrated robotic workcell. The effect of XKS activity on ethanol production was also evaluated by transforming the diploid strains containing the XI gene and each of the S. cerevisiae gene ORFs with pSUMOduo-RGStetHisXKS URA-selectable vector. Nine unique strains were isolated that grew anaerobically on xylose selective medium; two were found to no longer grow on glucose; seven were further evaluated for fermentation of alkaline peroxide-pretreated enzy-matically saccharified wheat straw hydrolysate. The selected PJ69-4 strains were analyzed and found to contain the following genes in the pOAD LEU-selectable plasmid: PIP2, IMG2, MAK5, VPS9, COX10, ALE1, CDC7, and MMS4 (all related to essential functions in the cell). 56 All strains successfully used glucose and xylose, consuming most of the glucose and a small amount of the xylose. Transforming the strains with an additional plasmid expressing the XKS gene did not improve anaerobic growth on xylose but improved glucose use and ethanol production on the hydrolysate, with three strains giving maximum ethanol production of 14.0 g/L. 56
Identification of Genes for Proteins That Bind Enzymes Needed for Xylose Utilization
Very little information is available in the literature on the binding of proteins to XL The introduction of Piromyces XI into yeast may expose the fungal enzyme to proteins and possible regulators that are not present in its natural environment. An automated two-hybrid interaction protocol was used 59 to find yeast genes encoding proteins that bind XI to identify potential targets for improving xylose utilization by S. cerevisiae. A pDEST32 vector reengineered for TRP selection and containing the Gal4-binding domain fused with the Piromyces sp. E2 XI ORF was used as bait with a library of LEU-selectable pOAD vectors containing the Gal4 activation domain in fusion with members of the S. cerevisiae genome ORF collection. Binding of a yeast ORF protein to XI activates two chromosomally located reporter genes in a PJ69-4 yeast strain to give selective growth. Five genes, ADH1, CSM4, APM1, RNR1, and YOR342C, were identified in the two-hybrid screen, suggesting that the proteins encoded by these genes bind to XI. 59
Four of these yeast proteins have known functions in the yeast cell, and one with the locus tag YOR342C does not have an assigned function. The four proteins with assigned functions are all enzymes that are critically important to the growth of the yeast cell. These may serve as potential targets for improving xylose utilization by S. cerevisiae. The effect of ADH1 overexpression was examined using an automated protocol to transform eight previously identified yeast strains that showed anaerobic growth on xylose. One of the transformants grew on wheat straw hydrolysate and consumed all available glucose, xylose, and arabinose. This strain will be used for further optimization of S. cerevisiae for cellulosic fuel ethanol production. Strains containing the other XI-binding proteins will also be used for further investigation.
The fact that different genes and thus different proteins were identified in the overexpression of yeast genes and the two-hybrid experiments is most likely because the two techniques were looking at different aspects of xylose utilization. The mating experiment was screening for genes that improved the process of xylose utilization, whereas the two-hybrid study was investigating genes that express proteins binding to XI, one enzyme important to the process of xylose utilization.
Genetic Engineering of Xylose-Using Yeast to Produce an Enzyme Catalyst for Biodiesel
The profitability of ethanol production from lignocellulosic biomass will be improved if high-value coproducts are also generated. Current processes for fuel ethanol production from corn starch yield substantial amounts of corn oil as a byproduct. The corn oil can be used for manufacture of biodiesel. The corn oil triacylglycerides are converted to fatty acid ethyl esters (biodiesel) and glycerol by transesterification with ethanol. One method of catalyzing this transesterification reaction is with lipase enzymes. 60 Use of lipases as bio-catalysts to accomplish transesterification would also help to decrease the costs associated with the traditional method of biodiesel production and to overcome some of the technical drawbacks,60,61 especially if large amounts of the lipases were expressed inexpensively in the yeast used in ethanol production processes in the biorefinery. An integrated biorefinery combining starch ethanol and cellulosic ethanol facilities may become more cost-effective if biodiesel is produced as a coproduct using lipase-catalyzed single-step column transesterification 62 with low-cost lipases expressed in large quantities in a recombinant yeast strain capable of cellulosic ethanol production.56,60
Advantages of using lipases include ease of product recovery, mild reaction conditions while minimizing energy consumption, and regeneration and reuse of the enzyme over several cycles. 60 In addition, purification of biodiesel can be accomplished on a resin to produce high-quality product that satisfies ASTM D6751 specifications.63–65 Although the cost of the enzymatic catalyst remains a hurdle compared with the less expensive chemical catalysts, the use of recombinant DNA technology to produce large quantities of lipases and the use of immobilized lipases may lower the cost of biodiesel production, while reducing downstream processing problems.60,66-68 The scripting of automated protocols and scheduling of PCR assembly steps on the robotic workcell have the potential to be used in an iterative fashion for production of any gene ORF. Rapid production of gene ORFs is essential for large-scale production of libraries of ORFs from full-genome sequences 46 or from systematically mutagenized optimized sequences based on a single-gene ORF.24,26
The Candida antarctica lipase B (CALB) gene ORF was produced using a stepwise oligonucleotide PCR assembly strategy followed by TOPO ligation directionally into pENTR D TOPO and LR clonase recombinational cloning into pYES2 DEST 52 vector for expression and evaluation of the lipase enzyme (Fig. 5).
69
The strategy previously described for PCR assembly of the XI gene ORF
42
involved a cloning step after each PCR step. The strategy described in Figure 5 eliminates the subcloning step and assembles the entire ORF in sequential PCR steps, so that the process is more rapid and readily adapted for the integrated robotic workcell. The Lyt-1 C3 variant gene ORF was added in frame with the CALB ORF using the automated PCR assembly and DNA purification protocol on the integrated robotic workcell. The fusion of the C3 variant of the Lyt-1 amphi-pathic peptide to the lipase potentially facilitates secretion and isolation of the expressed lipase outside the yeast cell for ready availability,
70
in this case for chemical attachment to a column resin for lipase-catalyzed biodiesel production. S. cerevisiae strains expressing CALB protein or CALB Lyt-1 fusion protein were first grown on 2% (w/v) glucose, producing 9.3 g/L ethanol during fermentation. The carbon source was switched to galactose for GAL1-driven expression of the CALB ORF, and the CALB and CALB Lyt-1 enzymes expressed were tested for fatty acid ethyl ester (biodiesel) production and were found to catalyze formation of fatty acid ethyl esters from ethanol and either corn or soybean oil. It was further demonstrated that a one-step-charging resin
71
specifically selected for binding to lipase was capable of covalent attachment of the CALB Lyt-1 enzyme and that the resin-bound enzyme catalyzed production of biodiesel.
Diagram of the stepwise assembly strategy used to construct the CALB gene ORF expression plasmids. Nine increasingly longer PCR amplicons, 6 at the 5’ end of the CALB sequence and 3 at the 3’ end, were created sequentially from 38 oligonucleotides (forward shown in yellow; reverse shown in tan) that included 36 consisting of 50 nucleotides, one consisting of 40 nucleotides and one consisting of 15 nucleotides for CALB 1 —38. Template 1 —26 and template 25—38 were combined using PCR to give the CALB 1 —38 construct (Top). PCR assembly and addition of five oligonucleotides containing the Lyt-1 sequence to the 3’ end of CALB 1 —38 to give CALB Lyt-1 1 —43 were performed on the robotic workcell. Purification of CALB 1 —38 and CALB Lyt-1 1 —43 was followed by TOPO ligation of each directionally into pENTR D TOPO and LR clonase recombinational cloning into pYES2 DEST 52 yeast expression vector (Bottom) for expression and evaluation of the lipase enzymes. Reprinted from Journal of the Association for Laboratory Automation, vol 16, Hughes, S. R.; Moser, B. R.; Harmsen, A. J.; Robinson, S.; Bischoff, K. M.; Jones, M. A.; Pinkelman, R.; Bang, S. S.; Tasaki, K.; Doll, K. M.; Qureshi, N.; Liu, S.; Saha, B. C.; Jackson, J. S.; Cotta, M. A.; Rich, J. O.; Caimi, P., Production of Candida antarctica lipase B gene open reading frame using automated PCR gene assembly protocol on robotic workcell and expression in an ethanologenic yeast for use as resin-bound biocatalyst in biodiesel production, pages 17-37, Copyright 2011.
Conclusion
High-throughput plasmid-based functional proteomic platforms that have the capacity to rapidly clone and express heterologous gene ORFs in bacteria and yeast and to screen large numbers of expressed proteins for optimized function are an important technology for improving microbial strains for biofuel production. Combined with rapid gene assembly and mutagenesis strategies on these platforms, gene ORFs can be synthesized, cloned, transformed into yeast strains, and screened to identify those that will give increased ethanol production, allow coproduction of biodiesel, enable use of biomass as a feedstock, and express valuable coproducts. Synthetic genes and the proteins with optimized functions they express form the basis of synthetic biology. Algorithms for combining the optimized genes to give the most efficient use of the improved properties are being developed. The approach for the past 10 years has been to overexpress proteins to enable microbes to perform functions that will allow more cost-effective production of biofuels. The next step is to generate stable strains containing the genes that overexpress the multiple proteins that were identified as having improved function. This will need to be coupled with technologies, such as Western blot analysis, high-throughput microscopy, mass spectrometry, gas chromatography, Raman spectroscopy, and microarray analysis. Adaptation of these technologies to a systems biology platform is possible to improve and screen any microbial strain. These techniques will allow tailoring microbial strains to use renewable feedstocks for production of biofuels, bioderived chemicals, fertilizers, and other coproducts for profitable and sustainable biorefneries.
Footnotes
Acknowledgment
Competing Interests Statement: The authors certify that they have no relevant financial interests in this manuscript and that any/all financial and material support for this research and work are clearly identified in the manuscript.