
Abstract
Robotic platforms are essential for production of large numbers of expression-ready plasmid sets to develop optimized clones and improved microbial strains for crucial bioenergy applications and simultaneous high-value peptide expression. Here we demonstrate a plasmid-based integrated robotic workcell, modified with a motorized vacuum filtration system, for performing fully automated molecular biology protocols, including assembly of mutagenized gene sequences, purification of PCR amplicons, ligation of PCR products into vectors, transformation of competent Escherichia coli, plating of recovered transformants, plasmid preparation, cloning, and expression of optimized genes. A library of genes encoding variants of wolf spider lycotoxin-1, a candidate bioinsecticide, was produced using PCR mutagenesis in an amino acid scanning strategy to generate a complete set of mutations across the lycotoxin-1 gene. The improved vacuum filtration system permits automated purification of PCR products. Methods for recovery and growth of bacteria containing plasmids with PCR inserts allow individual colony formation on a novel solid medium in a deepwell plate. Inserts are cloned into a bacterial vector to verify expression. These protocols form the core of a fully automated molecular biology platform that reduces the cost and time required to perform all operations. (JALA 2007;12:202–12)
Keywords
Introduction
Fully automated molecular biology protocols are essential to allow rapid cost-effective production of PCR-generated inserts for libraries, whether cDNA libraries or libraries of mutagenized clones, for incorporation into vectors, for plasmid recovery, and ultimately for insert expression. Although many molecular biology protocols are automated, 1 a robotic platform capable of carrying out the processes of PCR gene assembly and purification, cloning of PCR fragments into a vector, transformation of Escherichia coli, recovery of transformed cells, plasmid preparation, and protein expression in a continuous fully automated fashion has not been described previously.
An important application of such an automated platform is in the development and screening of optimized genes in high throughput for use in the production of improved commercial yeast strains to convert biomass to ethanol. 2 These strains are being engineered to express genes for hydrolysis and fermentation of cellulose and/or hemicellulose to ethanol. 3 At the same time, these strains can provide host capability for expression of high-value proteins and peptides. Genes for these proteins can be mutagenized and screened in high throughput to optimize the desired functional characteristics. The platform can be used for the optimization of genes for high-value peptides or proteins and the production of improved strains incorporating these genes and for numerous other applications in the biotechnology and pharmaceutical fields.
The liquid handler component of a plasmid-based functional proteomic robotic platform, described previously, 4 required development of a series of molecular biology protocols that have now been fully automated. Two significant improvements were made in the vacuum filtration block used for automated purification of mutagenized inserts: (1) providing motorized operation and (2) installing rotary cylinder fingers to maintain a vacuum-tight seal. A reliable vacuum system is essential for continuous unattended automated processing. Here we demonstrate the performance of these automated protocols and the use of the improved vacuum filtration block on an integrated plasmid-based robotic workcell to assemble and purify mutagenized open reading frame (ORF) inserts produced by an amino acid scanning mutagenesis (AASM) strategy, ligate these inserts at high efficiency into a topoisomerase (TOPO) cloning vector, transform these libraries in high throughput into E. coli, inoculate plates for plasmid preparation, recover the plasmids, and express the inserts, all in a fully automated fashion. We used a variation of the multiplex method 5 made possible by integration of a robotic colony-picking component onto an automated workcell platform. The data presented here show that we have successfully for the first time applied the integrated robotic workcell to the fully automated high-throughput construction and expression of a multiplex library of mutagenized ORFs for a representative peptide of potential commercial value.
Materials and Methods
Plasmid-Based Functional Proteomic Workcell
The design of the ProLink Express workcell assembled by Hudson Control Group has been described previously. 5 The workcell was modified to add a motorized Hudson VaryVac vacuum nest on the liquid handler to improve speed and accuracy of plate manipulation in the vacuum nest. This plasmid-based integrated robotic workcell was used to perform in a continuous operation fully automated molecular biology protocols for the PCR assembly of mutagenized gene sequences, purification of PCR fragments, ligation of PCR products into TOPO vectors, transformation into TOP 10 competent E. coli cells, plating of transformants, inoculation of cultures, and plasmid preparation. TrackLinks are connected through a Hudson PlateCrane for low-error parallel-plate operations.
PCR Synthetic Gene Assembly and Purification
A mutagenized histidine/enterokinase K/lycotoxin-1 (HIS/EntK/Lyt-1) library was generated using oligonucleotide synthetic gene assembly with oligonucleotides based on the Lyt-1 sequence from Lycosa carolinensis (GenBank Accession Number P61507). This 25 amino acid peptide toxin, lycotoxin-1, from wolf spider (L. carolinensis) venom was selected because it is effective against insects and not toxic to humans. 6 The starting sequence used was a mutagenized version of the Yan and Adams sequence 6 that had been optimized in a small-scale screen (data not shown) for insecticidal properties by random mutagenesis of the codons for the last four amino acid positions. This random mutagenesis strategy was described previously.4, 5 The last four codons preceding the stop codon in this mutagenized version of Lyt-1 (designated clone #59) were TTG TCG CCA TGG.
The sequence for Lyt-1 clone #59 was used in the AASM strategy in which each codon in the sequence was replaced by NNN, where N is any of the possible nucleotides. Each synthetic gene was produced from a 55-nucleotide forward oligonucleotide annealed to an 87-nucleotide reverse oligonucleotide with a 30-bp overlap. The forward oligonucleotide (55 nucleotides) contained the codons for the TOPO directional cloning signal, the 6× HIS tag, all four codons of the enterokinase K (EntK) site, and seven mutagenized codons of the toxin. The 25 mutagenized reverse oligomers (87 nucleotides) contained 3 codons of the EntK site and the NNN substitutions in the codons for each amino acid in the wolf spider toxin. The corresponding forward and reverse primers were 20-nucleotide and 19-nucleotide oligonucleotides, respectively. Oligonucleotides were synthesized by Sigma Genosys. Replacement of each codon in the long reverse oligomer for the 25 amino acids in the lycotoxin-1 peptide sequence potentially gives 64 reverse oligomers for each position for a total of 25 × 64 = 1600 possible synthetic genes.
Multiplexed PCR sets were prepared by mixing the long forward oligomer with each of the long reverse oligomers, including that of original optimized Lyt-1 clone #59. Three replicate sets, 1 set in each third of a 96-well plate were formed, consisting of the 25 mutagenized reverse oligomers plus duplicates of 2 of them, 2 clone #59 reverse oligomers, and 3 blanks, to fill 32 wells. The primers were added and PCR amplification was performed in a BioRad 0.2-mL well 96-well Hard-Shell PCR plate on the workcell in the thermal cycler using AmpliTaq and PCR reagents (Applied Biosystems) according to directions. Completed PCR reactions were purified on the workcell by adding the PCR reactions to the GENECLEAN II GLASSMILK/NaI reagent (Qbiogene) in a pre-assembled sealed ABgene 2.2 mL square well 96-well pyramid bottom deepwell plate. Reactions were mixed using the Variomag shaker on the workcell and passed through a Qiagen Turbo Plate in the automated modified vacuum nest by the liquid handler, held in the vacuum nest, washed three times with 1 mL NEW Wash (Qbiogene), and dried with fan-driven heated air. Bound purified PCR products were eluted after a 1-min delay with 30 μL of water into Matrix 2D-barcoded 0.75-mL collection tubes in a 96-tube rack in the vacuum nest lower position. The dynamic data link application written in the SoftLink control software for the vacuum filtration block permitted running of the vacuum nest from within the liquid-handler scripts originally written in the programming. 4
Multiplex TOPO Cloning and Transformation into TOP 10 Competent Cells
Mutagenized Lyt-1 clone #59 PCR products (4 μL) were transferred to a BioRad 0.2-mL 96-well Hard-Shell PCR plate on the workcell cold block at 4 °C for the TOPO ligation reaction mix with the Gateway adapted pENTR D TOPO vector (Invitrogen) according to directions, incubated in the thermal cycler at 23 °C for more than 5 h, and returned to the deck. At the same time TOP 10 E. coli cells (Invitrogen) were cultured in Luria-Bertanic (LB) medium (Teknova) overnight at 37 °C, with shaking at 400 rpm, then diluted approximately 1:15, divided into two ABgene 2.2 mL square well 96-well pyramid bottom deepwell culture plates, grown to log phase, centrifuged, and aspirated. Rubidium chloride transformation solution was added to the pelleted cells according to the directions in the Transformation & Grow Bacterial Transformation Kit (Qbiogene). The resuspended cells were incubated for 30 min at 4 °C and the TOPO plasmids added in another Hard-Shell PCR plate on the workcell. The transformation reaction was incubated for 30 min at 4 °C, moved to 23 °C for 30 min, centrifuged, and aspirated. Transformants were recovered in 900 μL of super optimal catabolite (SOC) medium (Teknova) in four ABgene 2.2-mL square well 96-well pyramid bottom deepwell culture plates for 7 h. Recovered transformants were spotted onto luria-bertani kanamysin (LB KAN) 25 solid medium plates (Teknova) for colony formation and selection. Colonies were grown for 20 h at 37 °C.
Multiplex Plasmid Preparation
The LB KAN 25 solid medium 96-well deepwell plates were flooded with 250 μL TB KAN 25 liquid medium (Teknova) and 20 μL were taken to inoculate four ABgene 2.2 mL square well 96-well pyramid bottom deepwell culture plates containing 1 mL TB KAN 25 medium to initiate plasmid preparation culture expansion. Twenty microliters from the four 96-well TB KAN 25 culture plates were used to broadcast inoculate four sets of four ABgene 10.0 mL square well 24-well round bottom deepwell liquid medium plates containing 10 mL TB KAN 25 medium. The plates were incubated in the Liconic incubator on the workcell with shaking at 400 rpm with a 1.5-mm orbit at 37 °C for 24 h. These sets were centrifuged and aspirated. The aspirated samples were used for Qiagen Turbo Minipreps according to kit directions. The 24-well cultures were processed in 96-well format into 4 sets of Qiagen Turbo Plates and Qiagen QiaPrep Plates from the Turbo MiniPrep Kit. The plasmids were eluted with elution buffer (EB) reagent (Qiagen) into Matrix 2D-barcoded 0.75-mL collection tubes in a 96-tube rack in the vacuum nest. These plasmid preparations provide readily accessible libraries of multiplexed mutagenized ORFs. Manual plasmid preparations to evaluate individual clones were performed according to Qiagen Turbo MiniPrep kit directions.
Plasmid Insert Sequencing
Plasmid inserts were sequenced using the ABI BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequencing was performed in 96-well format on an ABI 3730 capillary sequencer at Cogenics (Houston, TX).
In vivo Protein Expression Subroutine
Results and Discussion
The molecular biology protocols were performed in a continuous integrated fully automated process on a robotic workcell. A photograph of the Hudson Control Group ProLink Express workcell is provided in Figure 1. The day-by-day order of the protocols for the automated process and the plates required are presented schematically around the picture of the workcell. This platform was specifically designed to move insert libraries via LR clonase reactions into the novel small ubiquitin-like modifiers (SUMO) dual promoter vector (see Ref. 7 for parent vector) for expression in E. coli or yeast using an automated protocol. The improved vacuum filtration block provided consistent air-tight seals and allowed fully walk-away operation of the platform. No failures in the filtration or washing steps have occurred in the 5 months since the installation of the improved vacuum system.
Picture of ProLink Express workcell and day-by-day schematic outline of protocols and plate usage.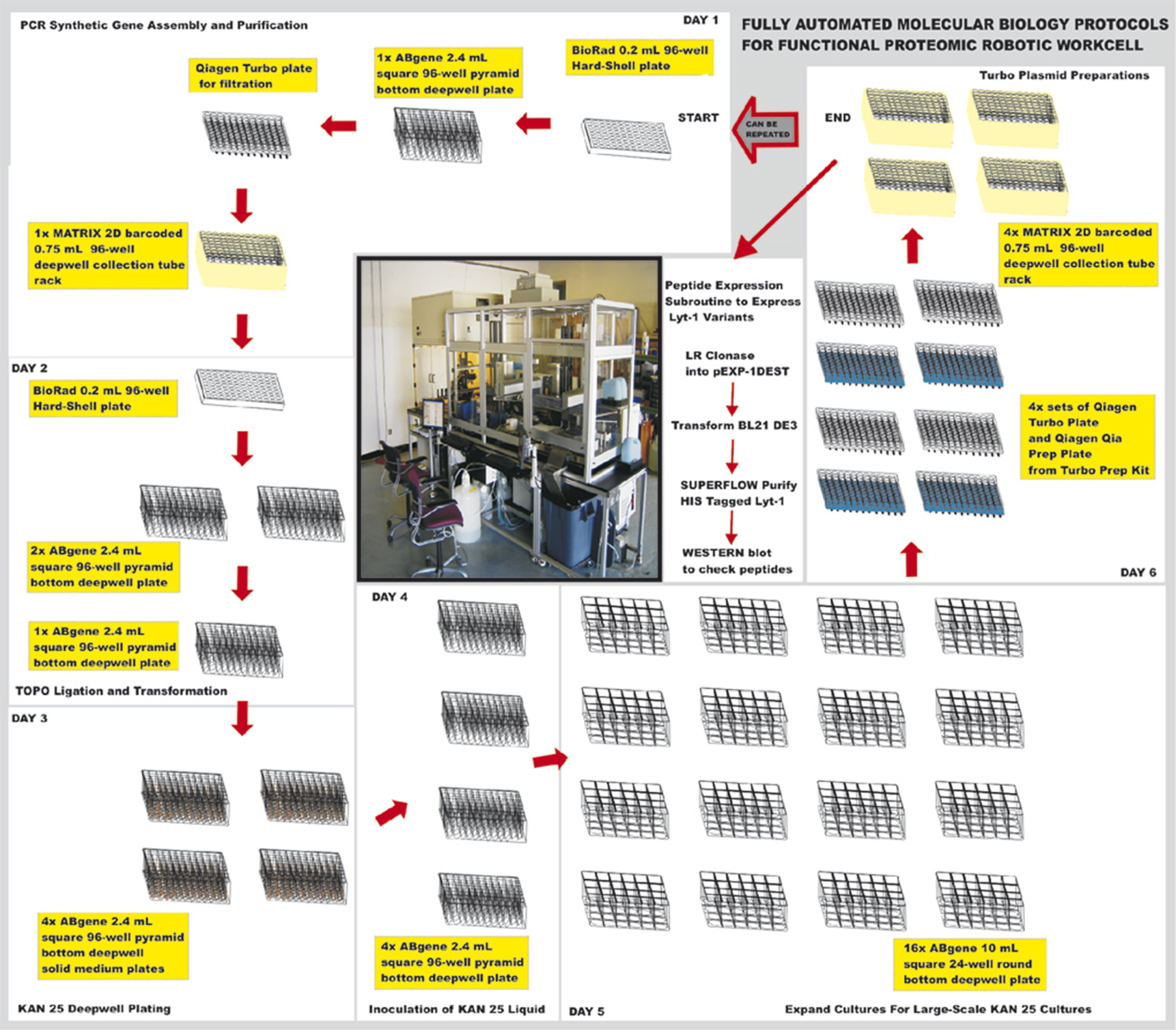
Oligonucleotides for AASM used in the PCR assembly of the synthetic genes were designed to generate all possible codons for each of the 25 amino acids in the lycotoxin-1 peptide from wolf spider venom (Fig. 2). Two long oligomers were synthesized. The 30 base pair overlap between the two long synthetic oligomers to form the PCR amplicon was engineered to be in the region of the 9 nucleotides coding for the last 3 amino acids of EntK in the forward oligomer and the 21 nucleotides coding for the first 7 amino acids of the toxin in the mutagenized reverse oligomer to which the forward oligomer anneals. The resulting 112 base pair amplicons code for a 6× HIS tag, an EntK site, followed by the sequences for each of the lycotoxin-1 mutants. The EntK sequence immediately preceding the toxin sequence is the site of cleavage by trypsin, which releases the toxin. In this case, the trypsin is from the BL21 DE3 bacterial cells. Another EntK sequence is present in the pEXP-1 DEST bacterial expression vector and the expressed insert can also be cleaved at that site. For the planned toxin expression in ethanologenic yeast, which will be fed to target insects, the trypsin to release the active toxin is present in the insect gut. No EntK sequence is present in the pSUMO DUO yeast expression vector. The only reverse oligomer not expected to anneal significantly to the forward oligomer is the one having a mismatch at the 3’ end caused by the NNN substitution there (BBCHTS 6-28-06K in Fig. 2). Each of the annealings would be filled on the first-round PCR cycle (Fig. 3A–C) providing a template for the 20 nucleotide short forward oligomer BBCHTS 6-28-06A (Fig. 2) and the 19 nucleotide short reverse oligomer BBCHTS 6-28-06B (Fig. 2). Further PCR amplification cycles with each template give a multiplex library of 1600 mutagenized amplicons (4 3 codon possibilities at each site × 25 codon sites).
Set of oligonucleotides for automated amino acid scanning mutagenesis (AASM) based on the original Lyt-1 mutagenized clone #59 ORF. Days 1–2: PCR amplification and purification of mutagenized ORFs on robotic workcell. (A) Placement of Matrix 2D plates in vacuum block with reconstituted oligonucleotides using blue jig to hold tubes. (B) PCR reaction on cold block on workcell deck. (C) PCR thermal cycler accessed by PlateCrane from Xantus track. (D) Binding of PCR products to purification reagents performed with shaking on Variomag. (E) Washing and drying of PCR products using automated vacuum filtration block. (F) Elution of PCR amplicons into Matrix 2D plate in lower position of vacuum block. (G) Sequence of plates as they appear in photographs. (H) Plate well designations for full set of amino acid scanning mutagenized amplicons. (I) Analysis of purified mutagenized Lyt-1 clone #59 amplicon set on 2% agarose gel. (J) Rear view illustrating the hardware modifications to the VaryVac vacuum nest system.
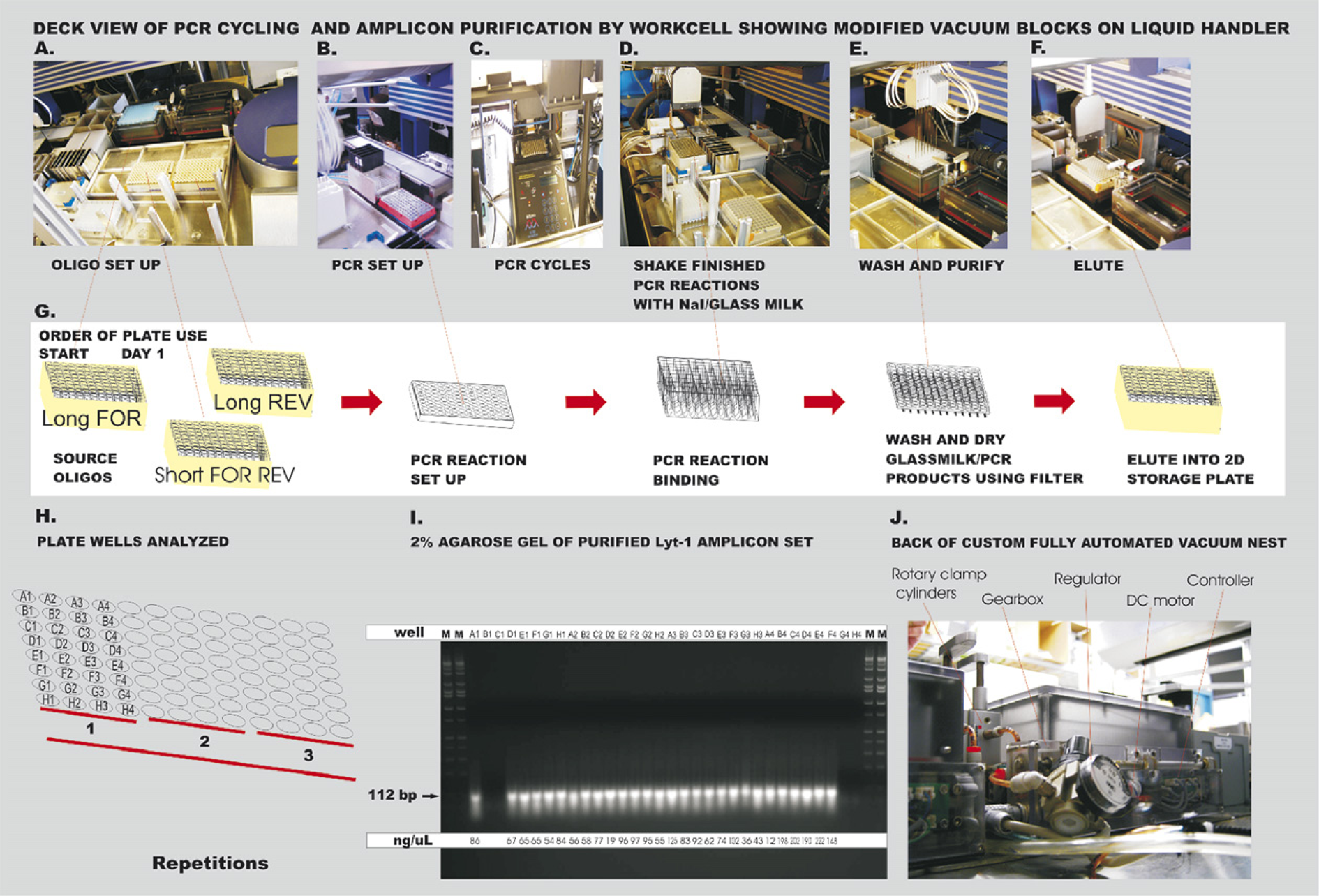
A protocol for efficient automated removal of contaminating free nucleotides and nucleases (Fig. 3A–F) from PCR amplicons is critically important to the process. Several methods exist but they do not allow recovery of very small DNA fragments (less than 300 base pairs). We found the GENECLEAN II methodology with Turbo Plate washing steps followed by drying and elution (Fig. 3D–F) could be readily adapted to the automated platform using the plates shown in Figure 3G. Indeed this methodology is capable of removing heparin from PCR products. 8 The combination of the improved vacuum filtration block and the use of the GLASSMILK reagent mixed with the PCR reaction allowed for binding and purification of the 112 bp PCR fragments. The first of the three replicate sets (wells A1 to H4 in Fig. 3) was analyzed completely (Fig. 3H gives the order of oligomers used and Fig. 3I depicts the agarose gel), and the analysis was repeated for the other two replicates (data not shown). The resulting 112 bp PCR amplicons were of sufficient purity and suitable concentration—most between 50 and 200 ng/μL as seen on a 2% agarose gel (Fig. 3I)—to allow TOPO cloning. These data demonstrated the ability of the procedure to recover small fragments that other methods had difficulty in retrieving. Use of the new automated vacuum block provided a significant advantage for filter purification of these fragments primarily as a result of the advanced design of the direct current (DC) motor that lifts the top nest portion of the vacuum block. The block top is rapidly moved into position by a gearbox and the filter plate in the top position is held securely in place using rotary clamp cylinders with aluminum “fingers” that press the plate down with sufficient force to form a tight, fail-proof seal with the rubber gasket on the top nest portion of the vacuum filtration block (Fig. 3J).
After free oligonucleotides were washed away from the bound double-stranded PCR amplicons and the purified amplicons were eluted (Fig. 3E, F), they were directionally cloned into the pENTR D TOPO vector. The CACC signal sequence placed at the beginning of the insert ORF is used to place the clone set with high efficiency into the pENTR D TOPO cloning vector (Fig. 4H–J) with the clones oriented in-frame for correct expression of the ORF. These TOPO ligations were transformed into TOP 10 E. coli cells made competent using rubidium chloride (Fig. 4A–G) on the deck of the robotic workcell just before transformation (Fig. 4K–O). Figure 4P shows the order of plates used. Because TOPO ligations have lower salt concentration, transformation is affected minimally by this preparation 9 and works well with the rubidium chloride method. The efficiency using freshly prepared competent cells 10 was compared on the robotic workcell with the efficiency using frozen cells 11 (Fig. 4Q). The Chung method 10 was found to be 20-fold more efficient for two different types of E. coli cells than the Hanahan method 11 (Fig. 4Q). This greater efficiency with rubidium chloride is better suited for the more complicated multiplex libraries where more transformations per well are needed. It was also easier to hold the fresh competent cells on the deck while the robot was performing the transformation than it was to hold the frozen competent cells. Part of the increased success was the improvement in piercing as a result of using the automated vacuum block fingers to hold the plate, allowing increased retrieval of ligation reaction for transformation (Fig. 4K). The process of preparing the cells on the deck was less expensive than buying frozen cells by a factor of approximately 6 for TOP 10 and 7 for BL21 DE3 transformations (Fig. 4R). Additionally, it was possible to carry out the automated transformation process at room temperature rather than purchase and thaw frozen competent cells.
Days 3–4: Preparation of TOP 10 competent cells and TOPO ligation on robotic workcell. (A) Sterile liquid fill performed by Hudson Micro 10 dispenser with automated 10-way valve. (B) Micro 10 sterile filling plates on track. (C) Liconic STX 44 autoincubator track fed with plates after sealing. (D) Dilution of overnight culture. (E) Dispensing diluted overnight culture evenly into 96-well plates for growth. (F) Pelleting cells in Ixion centrifuge after log phase growth. (G) Waste medium removed by track-based aspirator. (H) TOPO ligation setup on workcell cold block. (I) Reagents for TOPO ligation kept in cold block positions and transformation solutions kept in room temperature positions. (J) TOPO ligation incubated in PCR machine. (K) BioRad hard-shell PCR plate returned to vacuum block and held under rotary cylinder fingers to be pierced and pipeted into competent cells. (L) TOPO ligations held in vacuum block and pipeted into competent TOP 10 E. coli. (M) Cells with TOPO ligation reaction moved from cold block to the room temperature position. (N) Used BioRad hard-shell PCR plate sent down chute. (O) TOPO reactions transformed into TOP 10 competent E. coli recovered in SOC medium. (P) Order of plate usage. (Q) Study of the better efficiencies obtained with fresh competent cell usage. (R) Comparison of cost of freshly prepared versus purchased frozen competent cells.
The transformations were recovered in SOC liquid medium (Fig. 40) and selected on LB KAN 25 solid medium plates in 96-well format (Fig. 5A, B). The resulting minispreads on the solid medium plates, ranging from complete coverage to one or two colonies, were placed into a tower incubator at 37 °C (Fig. 5C). Colonies were flooded with TB KAN 25 liquid medium and used to inoculate a 96-well plate of 1.6 mL TB KAN 25 liquid medium for overnight growth (Fig. 5D–F). This plate was then used to inoculate four times four 24-well plates for plasmid preparation using four 96-well plate Qiagen Turbo MiniPrep kits to produce multiplexed plasmid libraries (Fig. 5G–L). The plasmid preparations were performed on the integrated Xantus liquid handler modified to incorporate a fully automated vacuum nest (Fig. 5M–U). Automation of this vacuum nest was accomplished by engineering a motorized lid with pneumatic aluminum fingers to push down on the plate when the lid is closed as shown in Figure 5S, T. This modification ensures that the plate forms a vacuum-tight seal even when material from the Qiagen solutions is deposited on the gasket over time causing a solid buildup. The pneumatic rotary cylinder fingers on the modified vacuum block exert sufficient force to keep the plates sealed tightly to the gasket despite the buildup. The pressure on the aluminum fingers can be adjusted to increase or decrease the force on the filter plates (Figs. 3J and 5T). This improvement/modification allows elution of the QiaPrep plates into Matrix tubes quickly and without loss of material or cross contamination in the second step of the Qiagen Turbo kit process.
Days 5–6: Large-scale plasmid preparation on robotic workcell. (A) Sterile medium- and solution-filled bottles attached to a 10-way valve to feed Micro 10 sterile track-based filler. (B) Inoculation of solid medium 96-well plates from SOC recovery cultures. (C) Incubator tower set at 37 °C for incubation of inoculated LB KAN 25 solid medium plates. (D) BioRad picker portion of integrated workcell. (E) Inoculation using flooded solid medium plates. (F) Pins of BioRad picker dipping into 96-well liquid TB KAN 25 liquid medium deepwell plates. (G) Filling 24-well plates with TB KAN 25 liquid medium for expansion. (H) Inoculation of 24-well plates on the deck of the Xantus using sterilized pipets. (I) Sealing of 24-well plates with porous tape. (J) Combination of PlateCrane and TrackLinks used to move plates with low error rates. (K) Loading and incubation of 24-well plates in Liconic incubator. (L) Position of all 16 24-well plates inside incubator. (M) Wash station eliminating use of disposable tips and stainless steel troughs for reagents for plasmid preparation. (N) Xantus deck setup for plasmid preparation. (O) Twenty-four-well plates brought to Xantus on track for producing glycerol storage stock plates. (P) Twenty-four-well plates pelleted before plasmid preparation. (Q) Twenty-four-well plates with pelleted bacteria aspirated. (R) Pellets receive plasmid preparation solutions from troughs and are shaken on Variomag shaker. (S) Blue QiaPrep plate placed into lower position of vacuum block. (T) White Qia Turbo plate placed into top position of vacuum block held down by fingers. (U) Plasmids eluted into Matrix 2D-barcoded plates and long-term storage cards produced. (V) Order of plate usage and identification of plates on the workcell deck. (W) Samples of multiplex plasmid from automated runs taken for BsrGI digestion. (X) BsrGI restriction analysis of plasmids on 2% agarose gel to observe multiplex inserts.
Transformation and plasmid preparation are portrayed in Figure 5A–U and the plates used are depicted in Figure 5V. Samples were collected in Matrix tubes (Fig. 5W). The multiplexed plasmids were restricted with BsrGI and analyzed for the presence of insert on a 2% agarose gel (Fig. 5X). The concentrations of most of the released inserts were between 15 and 41 ng/μL. The results show that only 3 out of 96 plasmids did not have inserts; 89.2% contained some types of insert. Inserts of two sizes were obtained, 180 bp (full length) and 160 bp, with 64 full-length inserts (61.4%) and 29 shorter ones. The shorter 160 bp inserts resulted from annealing of two long forward oligomers together instead of annealing of the long forward oligonucleotide to the long reverse oligonucleotide. This occurred due to the complementarity of the CAT sequence and the GAT sequence (when reversed) in the long forward oligomer.
Although the focus of this work was the automation of the molecular biology protocols, an evaluation of the individual clones was performed by taking several multiplexed wells, spreading them on LB KAN 25 plates and picking the colonies manually to isolate single plasmids for sequence determination. These colonies were cultured manually in 96-well plates and restricted with BsrGI (Fig. 6A). A representative colony was picked from each well and insert sequences were obtained for 90 of the 96 picked clones. Sequences for the inserts are shown in Figure 6B. None of the picked clones were mutagenized at the codons for the lysine repeats (see boxes in Fig. 6). Forty-five contained full-length inserts in the correct orientation; 36 of these were mutagenized versions of the Lyt-1 clone #59, and 9 were the original Lyt-1 clone #59. Two of the 36 mutants of clone #59 (clones 26 and 29) also had incorporation errors by Taq polymerase. Four had inserts in reverse orientation, 6 were truncated (1 also with a mutation of a codon), 3 ran past the end of the toxin due to an out-of-frame or incomplete stop codon (e.g., clone 38), and 13 had sequencing data that could not be deciphered. Nineteen did not use the long reverse oligomer in the PCR reaction, and the resultant annealing of two long forward primers produced an insert of approximately 160 bp as seen on the restriction analysis in Figure 5X. This single 96-well plate run yielded 36 plasmids containing full-length mutagenized genes ready to be moved into expression vectors for production of peptide variants.
Sequences of plasmid inserts from single colonies manually picked from the automated multiplex spreads, indicating amino acid positions that have been altered in each mutagenized insert and confirming insert lengths.
After these full-length plasmid inserts were cloned into the pEXP-1 DEST vector and expressed in BL21 DE3 bacterial cells, cell lysates were prepared and subjected to Western blot analysis. The lysates, each carrying a unique expressed sequence, were run on 16% acrylamide gels, blotted, and Penta HIS antibody was bound to the HIS-tagged fragments. It can be seen from Figure 7 that the mutagenized ORF inserts were successfully expressed. Three bands are observed for the HIS-tagged fragments resulting from ORF expression and subsequent cleavage by trypsin in the bacterial lysates (Fig. 7). The band at 8.9 kDa corresponds to expression of the full uncut ORF, HIS/V5/EntK/HIS/EntK/Lyt-1 (first 3 sequences from vector; last 3 from construct). The band at 6.4 kDa results from trypsin cleavage at the EntK sequence immediately preceding the Lyt-1 sequence to give HIS/V5/EntK/HIS/EntK (Lyt-1 is also produced in this cut because all inserts were full length, but it has no HIS tag and cannot be seen on the blot). The band at 6.0 kDa corresponds to HIS/EntK/Lyt-1. All lysates show this band, indicating that mutant Lyt-1 toxin is released from each. The lysate with clone #59 (indicated as wt in Fig. 7) shows only this band. The lysate containing clone #29 has large amounts of uncleaved expressed ORF. The clone #25 lysate primarily shows the band at 6.4 kDa. Further studies are being conducted to determine the effectiveness of each of these lycotoxin-1 variants as an insecticide. The production of protein was carried out to obtain an estimate of the cost of this automated process using AASM versus that using the random error-prone PCR mutagenesis (EPPM) process.
Western blot analysis depicting fragments released after expression of mutagenized inserts in BL21 DE3 bacterial cells and cleavage by the bacterial trypsin. All inserts show a band at 6.0 kDa, indicating mutant lycotoxin-1 is released from each.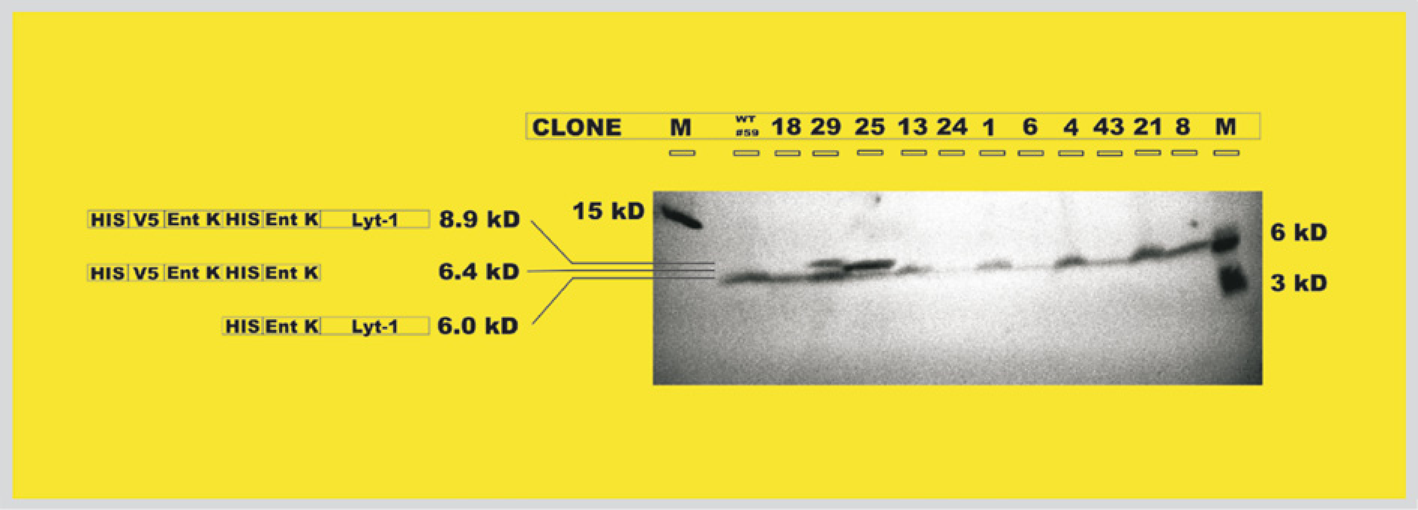
The sample size, time, and cost requirements for the fully automated AASM process carried out on the workcell using the ABI kit for PCR compared to the requirements estimated for the automated EPPM process using the GeneMorph II kit to produce the same number of mutations are given in Table 1. In the AASM process, all possible mutations are produced at every codon in the ORF. The EPPM process (1 mutation every 1000 bases) requires many more samples to obtain the same number of mutations and all codons may not be mutagenized because it is a random process. The EPPM process requires more than 30 times as many sample wells as the AASM process. Overall, the EPPM process takes 6 times longer and costs approximately 16 times more than the AASM process.
Summary of the sample well, time, and cost requirements for automated AASM (actual) as carried out using ABI kits for PCR steps compared to the requirements for automated EPPM (estimated) using GeneMorph II kits (1 mutation per 1000 bases)
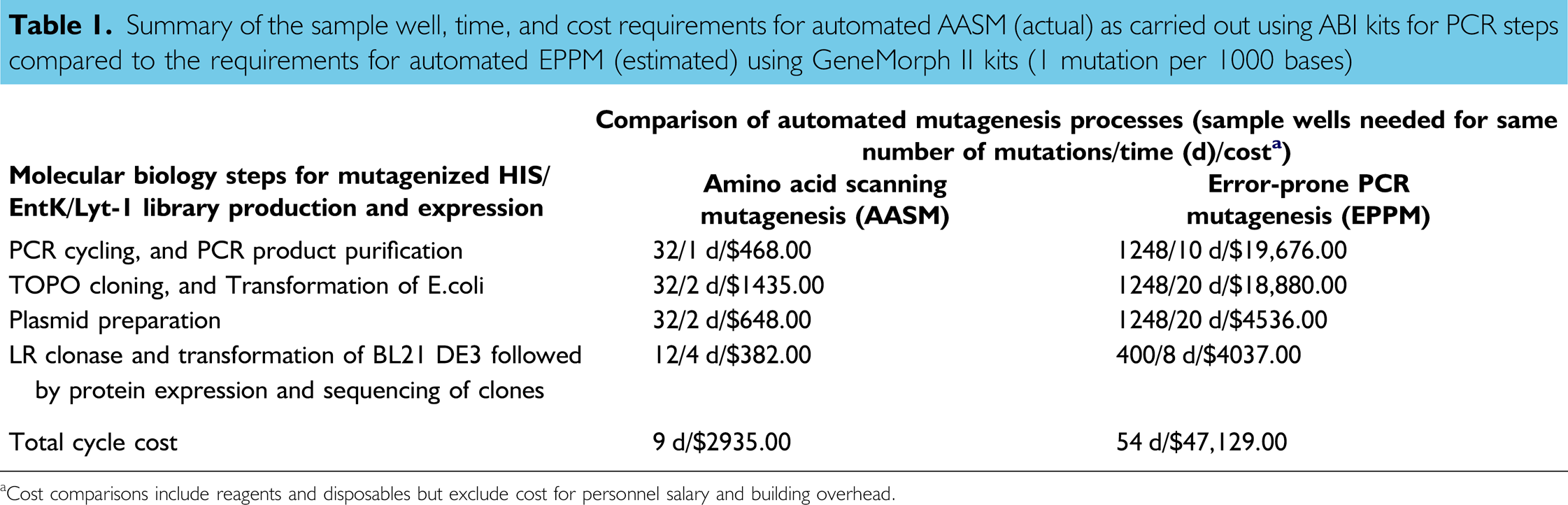
Cost comparisons include reagents and disposables but exclude cost for personnel salary and building overhead.
Conclusion
To construct a library of variants of the lycotoxin-1 gene, a core set of fully automated molecular biology protocols was developed and performed on a plasmid-based integrated robotic workcell. Two significant improvements were implemented in the design of the vacuum filtration block on the workcell, motorized operation, and addition of rotary cylinder fingers, to permit filtration and washing of plates in a fail-free operation. The entire process from PCR assembly of mutagenized genes to production of plasmid libraries was conducted in a continuous fully automated operation. All steps in the process, including purification, ligation, transformation, and plasmid preparation were carried out with high efficiency resulting in 89% of the plasmids containing inserts. Of the 90 plasmids sequenced, 36 were ready to be used to move the inserts into expression vectors. Twenty-four of these were cloned into a bacterial expression vector and expressed in BL21 DE3 cells. All mutagenized ORF inserts released a lycotoxin-1 variant peptide. These results obtained with an insecticidal peptide gene demonstrate that the robotic workcell platform can be used for the fully automated preparation of a multiplex library of mutagenized ORFs. A comparison of automated ASSM and EPPM shows that the EPPM process requires over 30 times as many sample wells, takes 6 times longer, and is more expensive by a factor of 16 than the AASM process to obtain the same number of mutations.
Footnotes
Acknowledgments
The authors would like to thank Jennifer Steele for her help in sequencing the lycotoxin mutants. They wish to acknowledge the assistance of Robert C. Bobenchik, John J. Celecki, Raymond L. Dailey, Claudio Goncalves, Valentin Gorbunov, Vladimir Gorbunov, Rosemarie T. Kramer, Debra A. Peterson, Bruce M. Pierpont, P. Robert Reed, and Igor Zolochevskyy at Hudson Control Group in the design and construction of the modified vacuum block. The authors would also like to thank Watson Chau for help with plasmid preparations and sequencing and would like to express their appreciation to Karen Hughes for editing and formatting this manuscript.