
Abstract
A Hudson Control Group, Inc. ProLink Express™ robotic workcell to conduct plasmid-based functional proteomics is being developed for optimization of protein open reading frames (ORF). The initial phase of this project is to design and assemble a Xantus liquid handler from Sias, Inc. modified by Hudson so that a workcell track component can be placed within the Xantus® gripper tool work area. The liquid handler is designed to produce plasmids using the Qiagen Turbo® plasmid preparation kit. This design allows processing of up to four 96-well plates in one run. The procedure eliminates disposable tips and provides an advanced wash system to prevent cross contamination. To evaluate liquid handler operation, a mutagenized cellulase F ORF plasmid library was prepared from wild-type cellulase F (Chen, H.; Li, X.-L.; Blum, D. L.; Ximenes, E. A.; Ljungdahl, L. G.CelF of Orpinomyces PC-2 has an intron and encodes a cellulase (CelF) containing a carbohydrate-binding module. Applied Biochemistry and Biotechnology 2003, 105–108, 775–785; Li, X.-L.; Chen, H.; Ljungdahl, L. G. Two cellulases, CelA and CelC, from the polycentric anaerobic fungus Orpinomyces strain PC-2 contain N-terminal docking domains for a cellulase-hemicellulase complex. Applied and Environmental Microbiology 1997, 63(12), 4721–4728) using a novel Invitrogen Gateway® cloning strategy. For the automated reproducibility run, the average yield of plasmid was 5.35 μg per well from 1.347 mL of starting culture. Four plates were processed automatically on the liquid handler in 374 min compared to at least 441 min for the same plate operations performed manually. The quality and quantity of plasmids prepared on the liquid handler made the implementation of the following workcell protocols possible: DNA sequencing, in vitro transcription/translation, and transformation of bacterial and yeast strains for protein expression.
Introduction
The field of proteomics is moving beyond identification of proteins by mass spectroscopy of tryptic peptide fragments or microsequence of proteins isolated from 2D gels. Numerous methods for obtaining and identifying proteins have been de-veloped, 4 –5 but these methods are often limited in their ability to detect proteins present at low levels, and frequently do not provide enough information to clone the genes. In an attempt to overcome these limitations, attention has been turned to plasmid-based functional proteomics. This technology centers on the production of full-length cDNA libraries as a source of plasmid-based clones for identification of protein function. These clones can be used in mutagenesis strategies for open reading frame (ORF) optimization to develop improved strains and cell lines.
Plasmid-based functional proteomics requires rapid plasmid preparation methods to obtain adequate quantities of high-quality plasmid DNA to conduct all the required steps in the process from plasmid production to protein assay. Because the plasmid libraries are composed of several thousand unique genes, automation of the process is essential. To date, a number of automated systems have been de- veloped, 6 –10 but no single automated system is available that integrates the entire process into one continuous programmed sequence, from creation of plasmid libraries to functional testing of expressed proteins. Many cloning strategies have been used, 11 –18 however, cloning strategies for high-throughput expression and screening on an automated workcell are not currently available. The ideal system would be capable of producing full-length cDNA libraries, colony picking, isolating plasmid DNA, transforming yeast and bacteria, expressing protein, and performing appropriate functional assays. Such an automated system will clearly require the integration of different equipment and instruments with the desired capabilities. One of the most important pieces of equipment is a precise and reliable liquid handler.
We are currently building an integrated plasmid-based proteomic workcell to automate all the required tasks from library creation through functional testing for large sets of clones. In this work, we highlight the functionality and reproducibility of a modified Xantus liquid handler that will support the proteomic workcell procedures. The liquid handler was used to isolate plasmid DNA from bacterial cultures in 96-well plates. As an additional proof of concept, a mutagenized celF library was screened. celF plasmid DNA was used for sequencing, in vitro transcription/translation, and transformation of bacterial and yeast strains followed by assays of the expressed proteins to demonstrate that plasmids prepared on the liquid handler are capable of producing active proteins for functional analysis.
Materials and Methods
Liquid Handler Assembly and Testing
Assembly and integration were performed by Hudson Control Group, Inc. using Sias, Inc. components. The plasmid-based functional proteomic robotic workcell requirements for the liquid handler involved modifying the deck plates of a 1.5 m long Xantus liquid handler (Sias). The Xantus liquid handler has several I/O type USB ports providing connectivity for additional instrumentation and is controlled by custom Xantus Application Software (XAP). To perform the Qiagen Turbo® Miniprep Kit, several equipment additions were made to the five stainless steel Xantus deck plates. These deck modifications included a reagent circulating system and reservoirs for the liquid reagents so a constant supply of fresh reagent can be pipeted. Wash solution reservoirs provide bleach and ethanol solutions for cleaning and sterilizing pipets after each use. Temperature of the cold reagent reservoir is maintained with coils attached to a Julabo F12 chiller circulating water bath. A modified Qiagen QIAvac 96 vacuum filtration block, typically used for the 96-well filter plates associated with the QIAprep 96 Turbo Miniprep Kit, was installed. An Ixion microplate centrifuge to pellet bacterial cultures and a Variomag® plate shaker with custom interface for resuspension of cells were added. The modifications also involved a sophisticated vacuum pump system with a custom regulator that controls the pressure delivered and the fluttering of the vacuum applied. The liquid handler has a custom drying station that can remove any residual wash solution completely with a constant supply of heat and a continuous draw of air supplied by a small fan. Track elements leading from the workcell were integrated to the deck of the Xantus to position incoming deepwell culture blocks for processing by stopping them at StopLink plate positioners.
The reliability and reproducibility of the liquid handling system were tested by performing the entire plasmid preparation procedure multiple times. To determine the precision and accuracy of the liquid handling component, the final yield of plasmid was measured across 96 wells on four different plates. The efficiency of the plasmid preparation protocol was tested in the same manner by evaluating the quantity and quality of plasmid DNA. Finally, to ensure that there was no well-to-well carryover by the liquid handler, four different plasmids, pUC19, pEXP-1 DEST GUS, pEXP-1 DEST CelF #5, and pEXP-1 DEST CelF #62, were inoculated in a 16-well grid pattern and the absence of cross contamination confirmed by sequencing and restriction enzyme digestion.
Plasmid Preparation and Analysis
Plasmids were prepared on the liquid handler using the QIAprep 96 Turbo Miniprep Kit (Qiagen) with 1.347 mL of bacterial culture grown in 96-well deepwell plates. The manufacturer's protocol was followed except that in the final step the elution buffer (EB) was divided into two aliquots of 75 μL for the mutagenesis run or two aliquots of 100 μL for the automated reproducibility run instead of one 150-μL aliquot. Spectrographic analysis of plasmid samples was performed using the NanoDrop ND-1000 fiber optic spectrophotometer (NanoDrop Technologies). Restricted plasmid preparations were run on 2% agarose gels with ethidium bromide (Sigma) added to visualize the DNA. The gel was photographed with an Alpha Imager 3400 using software version IS-3400 V 3.1.2 for Windows 2000/XP. Plasmids were sequenced using a MJ Tetrad PCR machine model PTC 225 with the Big-Dye Terminator V3.1 (Applied Biosystems) according to the manufacturer's instructions and run on an ABI 3730 DNA Analyzer (Applied Biosystems).
Cellulase F Mutagenesis Library
A mutagenized celF library was generated by synthesizing oligonucleotides in which the last four codons of wild-type celF were replaced with the NNR motif using the following PCR primers:
Forward primer BBCHTS 11-5-04B 5′- CACCATGAAAATTTTACTTTTTGCCAGTATTCT TAGTTTTGG-3′ Reverse primer BBCHTS 11-2-04A 5′ -TTAYNNYNNYNNYNNAGCATTTCTTAATAATT GAACGAAATAATCG-3′
PCR was performed using AmpliTaq® and PCR reagents (Applied Biosystems) with celF cDNA (GenBank Accession Number U97154) from the anaerobic fungus Orpinomyces PC-2 19 as template (100 ng/μL) according to the directions. The resulting PCR products were run on 2% agarose gels (Sigma). The gel was placed on a UBI 320 nm UV light box, and the 1364 bp mutagenized celF fragments were cut out and then purified using the Qbiogene GENECLEAN® II Kit according to the directions provided.
TOPO Ligation into pENTR D TOPO
A 4-μL aliquot of the purified mutagenized celF fragments was used in a TOPO ligation with the Gateway® adapted vector pENTR D TOPO for directional cloning and transformed into TOP 10 cells according to the manufacturer's instructions (Invitrogen). Resulting colonies were picked with a BioRad VersArray™ picker into ABgene deepwell blocks holding 1.6 mL TB KAN medium (Teknova per well). After growth, the cultures were run through the four-plate liquid handler process, and the plasmids, glycerol stocks, and long-term storage cards were prepared and collected.
LR Clonase Reactions
After the set of pENTR D TOPO 1364 NNR plasmids was collected, LR clonase reactions were performed to move the mutagenized insert into pEXP-1 DEST (Invitrogen) for in vitro expression in bacterial lysate containing T7 polymerase, pDEST 17 (Invitrogen) for in vivo expression in bacteria, or pYES2 DEST 52 (Invitrogen) for in vivo expression in yeast. LR clonase reactions were carried out according to the directions in the Invitrogen kit, and 4 μL of the resulting reactions was transformed into TOP 10 chemically competent Escherichia coli cells. After growth, the resulting colonies were picked by the BioRad picker and inoculated into a deepwell plate containing either 1.6 mL LB AMP (Teknova) or TB AMP 50 medium (Teknova) and incubated for 18 or 24 h. The resulting DEST expression vectors in TOP 10 were processed using the Xantus liquid handler protocol for plasmid preparation.
Bacterial Transformation and In Vivo Cellulase F Expression
The pDEST 17 library plasmids with mutagenized celF prepared on the liquid handler were transformed into BL21 DE3 using competent cells from Stratagene according to the manufacturer's directions. The colonies were picked for 2 h into 1.6 mL TB AMP 50 medium at 37°C, induced with 20 μL of 100 mM isopropyl-β-D-thiogalactopyranoside (IPTG) (Novagen), and the cultures incubated for 20 h. Lysates were prepared with B PER II® Bacterial Extraction reagent (Pierce), mixed with 2 × tris glycine loading buffer (Invitrogen) containing β-mercaptoethanol (BME) (Sigma), and heated at 95° C for 5 min. A sample of 20 μL was used for Western blot analysis, and 3 μL was spotted directly on an azo-carboxymethyl cellulose (azo-CMcellulose).
In Vitro Transcription/Translation of Cellulase F
A 5-μL aliquot of the pEXP-1 DEST CelF NNR plasmids prepared on the liquid handler was used for in vitro transcription/translation reactions with the T7 lysate from Roche Molecular Kit RTS 100 to generate recombinant protein. Half reactions at 25 μL were performed instead of the full reaction at 50 μL as directed in the kit. Reactions were incubated for 6 h at 30° C. Proteins obtained from in vitro transcription/translation reactions were purified by adding 50 μL Ni Superflow beads (Qiagen) to 50 μL of reaction mixture to bind the HIS-tagged CelF. The beads were incubated with shaking at 4°C overnight after addition of 1 × PBS (Cellgro) to 400 μL, then centrifuged and washed with 400 μL 1 × PBS, followed by centrifugation and elution of the bound CelF with 30 μL 250 mM imidazole (Sigma) in 1 × PBS. Eluate of 20 μL was taken for Western blot analysis, and 3 μL was spotted on azo-CMcellulose plates. For the reproducibility runs, 20 μL of the transcription/translation reactions was used directly for Western blot analysis, and 5 μL of the reactions was spotted for azo-CMcellulose plate analysis.
Yeast Transformation and In Vivo Cellulase F Expression
The pYES2 DEST 52 plasmids prepared on the liquid handler were moved into yeast INVSc 1 using Qbiogene EZ-Yeast Transformation Kit. On completion of the transformation, the ura3 plasmid-containing yeast transformations were entirely plated to CM-URA glucose 100 mm circular plates (Teknova) for recovery. Transformants were identified after 2 days of growth at 30° C, then picked by hand into 1.6 mL CM-URA galactose medium (Teknova), and incubated for 2 days at 30°C. Lysates were prepared using 1.6 mL of culture centrifuged for 10 min at 2500 rpm and 4°C. After centrifugation, the pellets were treated with 50 μL Y-PER® -S Yeast Protein Extraction reagent (Pierce) from which lysates were prepared, and 3 μL was spotted on an azo-CMcellulose plate.
Western Blot Procedure
Western blot analysis was performed by mixing 20 μL of sample with 20 μL of 2× loading buffer containing tris glycine and BME. The samples were heated at 95°C for 10 min, then loaded onto 1.5-mm thick 16% tris glycine 15-well SDS-PAGE gels (Invitrogen). The gel was assembled into an X-Cell® Novex box and run in 1 × tris glycine running buffer. It was removed and transferred to PVDF cut sheets (Invitrogen). The transfer took place in the Novex transfer rig with 1× transfer buffer (Invitrogen) for 10 h at a constant 200 mA. Chromogenic Western blots were performed with the Western Breeze kit (Invitrogen) according to the manufacturer's directions using a Qiagen PENTA HIS antibody resuspended in 1 mL of 1 × PBS (Cellgro). Reconstituted antibody of 30 μL was used in the primary antibody step. Western blot images were captured using the Alpha Innotech 3400 and analyzed for the amount of full-length CelF expressed. The AlphaEaseFC software for Windows 2000 was used to determine band density compared to the known molecular weight markers from the Qiagen 6 × HIS-tagged set treated in the same fashion as the CelF samples.
AZO-CMcellulose Plate Assay
Plates were prepared by dissolving 2 g AZO-cellulose from Megazyme in 800 mL Milli-Q water. The pH was adjusted to 4, 5, 5.8, 7, and 8, then 15 g of agar (Sigma) was stirred in, and water added to 1 L. After autoclaving at 121°C for 15 min on the liquid setting, the solution was again stirred before being poured into the Omnitrays from NUNC. The plate was allowed to cool and solidify for 1 h. Matrix multichannel pipets with a well jig were used to spot 3 μL of lysate. Plates were inverted and incubated at 37°C for 18 h.
Results and discussion
Liquid Handler for Plasmid Preparation
The 1.5-m Xantus liquid handler was selected for incorporation into our plasmid-based functional proteomic robotic workcell because of its rugged deck layout constructed of stainless steel plates, large working area, and compact footprint. A microgear pump drives each pipet of the eight-span pipet set, allowing continuous flow and operation in a sterile pump housing. The liquid handler uses sterile water as the system fluid. Pipet tips are sterilized between each use in a 10% bleach wash, followed by a 70% ethanol rinse, and finally a water rinse. This eliminates disposable tips and the need for space to store them.
The liquid handler layout (Figs.
1
–3) is designed to allow Hudson Control Group,
Inc. TrackLink and StopLinks to bring in two ABgene polypropylene 96-well deepwell
culture blocks, containing 1.6 mL bacterial cultures, within the Xantus gripper
workspace. This format was selected because the Ixion centrifuge (Fig. 1) on the Xantus deck can
accommodate two deepwell plates at a time. This two-plate process is run twice for a
total of four 96-well plates. The plates are incubated for 18 or 24 h at 37° C. These
processes are part of a workcell into which the liquid handler is integrated. The XAP
written by Hudson to prepare plasmids was designed with the intention of initiating
the appropriate sequence of operations in the workcell scheduling software, SoftLink,
from Hudson Control Group, Inc. Figure 4 provides an overview of all protocols optimized for the workcell
using plasmids prepared on the liquid handler. The boxes shaded in aqua in
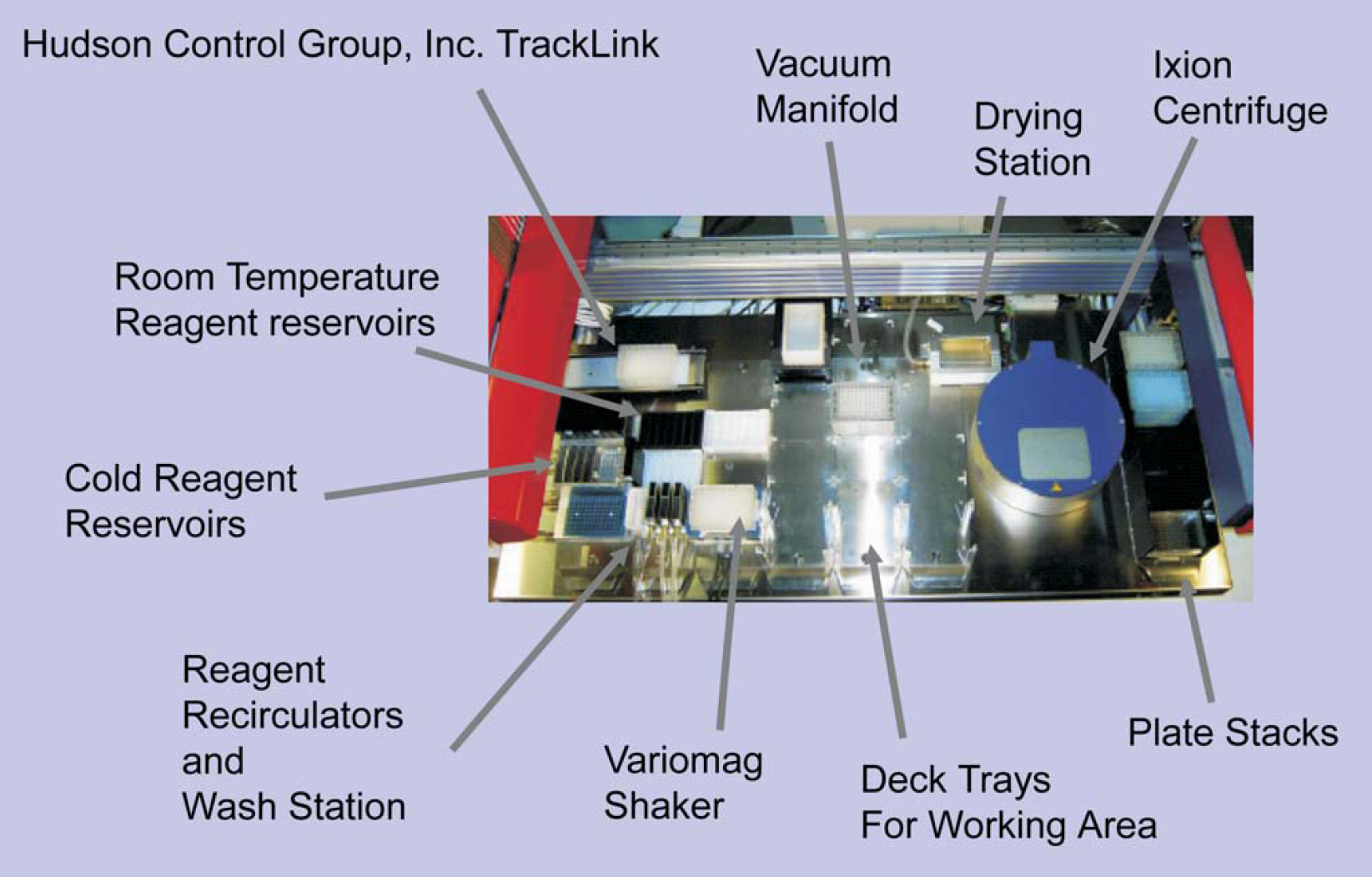
Arrangement of equipment added to the liquid handler deck. Diagram indicates incoming track element from workcell

Robotic components of the liquid handler are the gripper arm and the eight-span pipet arm. Pipets are Teflon coated and sterilized between sample delivery to eliminate need for disposable tips.
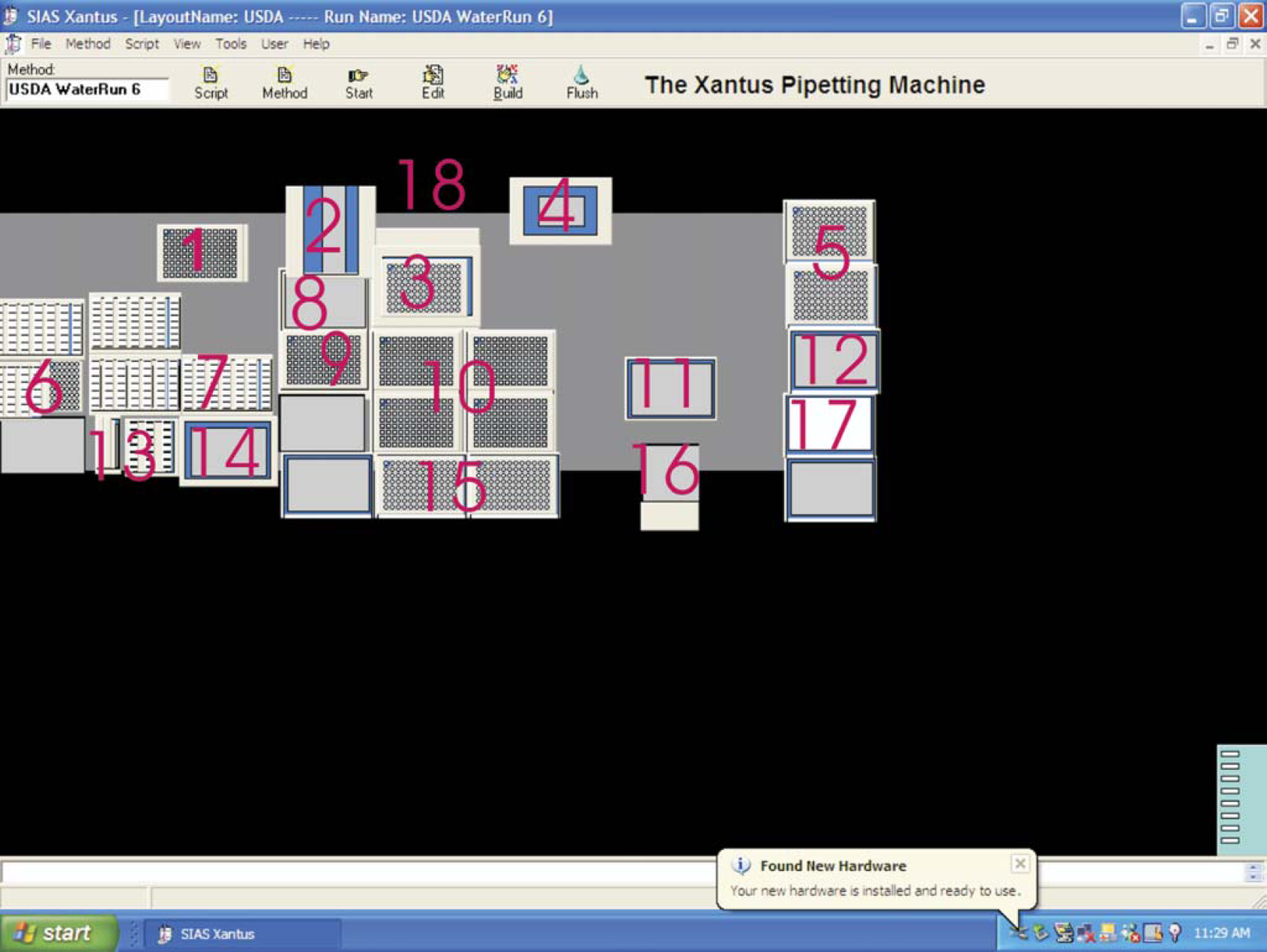
Deck positions on the liquid handler as identified in the XAP
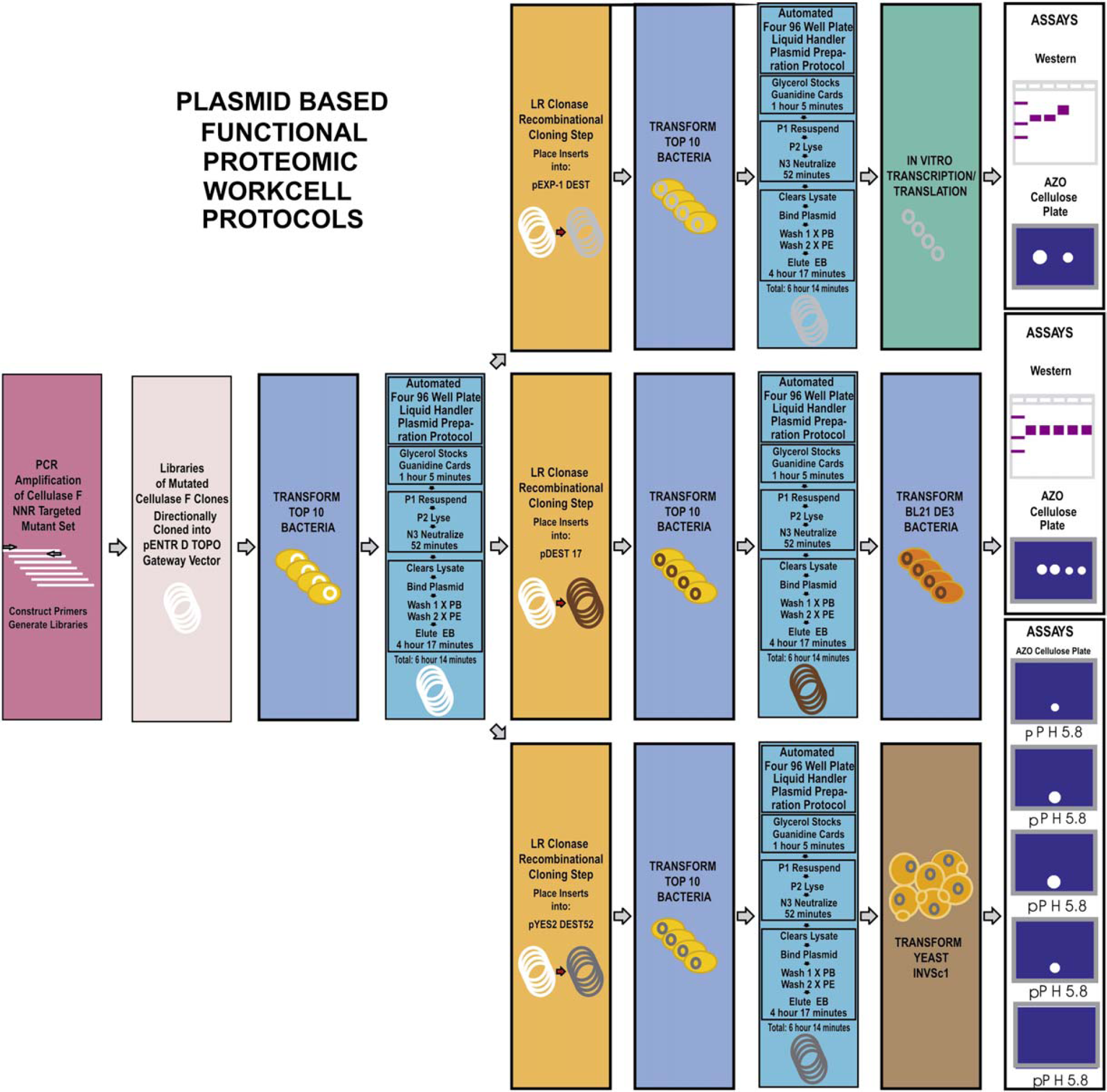
Overview of molecular biology protocols optimized for the workcell with the plasmid preparation operations on the liquid handler indicated by the boxes shaded in aqua
The XAP plasmid preparation protocol is initiated when the cultures have grown to log phase, and they are transported to the deck of the liquid handler on Hudson Control Group, Inc. TrackLink (Fig. 1) within the Xantus gripper workspace. To prepare for long-term storage of samples from each of the deepwells, 2D bar-coded 1.4-mL Matrix plates (Fig. 5) are moved into the glycerol stock positions (Fig. 3 position 10) on the deck and filled with 600 μL of sterile 50% glycerol. Omnitrays with clean guanidine cards (Fig. 5) are placed in the Omnitray positions (Fig. 3 position 9) of the deck with lids removed (to lid holding spot, Fig. 3 position 2). The first two deepwell culture plates (Fig. 5) are transported to the Xantus deck, with the first plate in the track access position (Fig. 3 position 1). Aliquots of 253 μL of the bacterial cultures are taken up into the pipets, and 3 μL is spotted onto DNA storage cards (Whatman paper impregnated with guanidine) held in delidded Omnitrays in the Omnitray position on the liquid handler. The remaining 250-μL aliquot of culture in the pipets is dispensed into 600 μL of 50% in a glycerol stock Matrix plate (tips washed between each eight-set culture aliquot delivery) and each plate moved to the outgoing stack (Fig. 3 position 12) when completed. The Omnitrays holding the guanidine cards are lidded and also moved into the outgoing stack (Fig. 3 position 17).

Examples of plates used for plasmid preparation
The first of the deepwell blocks on the track access position (Fig. 3 position 1) is then moved by the gripper (Fig. 2) into position A of the Ixion centrifuge (Fig. 3 position 11). Next, the second deepwell culture block is moved by the gripper into position B of the centrifuge. Both are centrifuged for 10 min at 2000 rpm. After centrifugation, the blocks are moved back to the track access position and taken by the track to a medium removal area. After the used culture medium is removed, the deepwell blocks with pelleted bacterial cultures are returned to the deck of the Xantus with the first plate arriving at the track access position (Fig. 3 position 1).
The gripper then moves the first deepwell block with pellets to the Variomag® shaker (Fig. 3 position 14) and, with a “swish and swirl” technique devised for this workcell, resuspends the pellet in 250 μL of P1 obtained from a cold reagent reservoir (Fig. 3 position 6). While P1 is being aspirated, the plate in the Variomag position is shaken at 1100 rpm resulting in a swish and swirl technique that completely resuspends the bacterial pellets. A 250-μL amount of P2 from a room temperature reservoir position (Fig. 3 position 7) is then added to each well of the first deepwell block and shaken for 30 s to mix (1100 rpm). The block is moved to a holding position (Fig. 3 position 10) on the deck and held until the second plate is processed. The process with P1 and P2 is repeated for the pellets in the second deepwell block.
The first plate is brought back to the Variomag position, and 350 μL of N3 is added to each well of the plate from the room temperature reservoir (Fig. 3 position 7) and shaken at 1100 rpm for 30 s. The QIAvac 96 filtration block is assembled (Fig. 3 position 3) with a blue QIAprep plate from the filter plate stacks (Fig. 3 position 5) in the lower holder position, and the Lucite top is placed over the QIAprep plate with the white TurboFilter plate in the top position. The resulting bacterial lysates are neutralized (generating visible debris), placed into the TurboFilter plate, and pulled through slowly with a vacuum of approximately 300 mTorr for 10 min to ensure that all the lysate is cleared by the TurboFilter plate. Between each lysate movement into the Turbo plate is a wash step. The TurboFilter plate is discarded in the waste chute (Fig. 3 position 16).
The Lucite top of the QIAvac 96 vacuum block is put in the rear park position (Fig. 3 position 18), and the QIAprep filter plate with cleared aqueous solution containing plasmid is placed into the Lucite top of the QIAvac 96 filtration block. The entire Lucite top is reassembled onto the base that now has the modified receiver piece to evacuate all waste wash liquids into a waste receptacle. After the top has been placed onto the QIAvac 96 bottom with the QIAprep plate and aqueous solution in the top, the vacuum is turned on and the cleared lysate is drawn very slowly through the QIAprep filter plate at approximately 200 mTorr for 5 min to ensure maximal binding of plasmid DNA after neutralization.
The QIAprep filter plate is then washed with 800 μL of PB solution from a sterile stainless steel reservoir in the recirculated reagent positions (Fig. 3 position 13), which ensures a fresh supply of wash solution for removal of residual contaminating proteins, including any RNase A from solution P1 that might interfere with in vitro transcription/translation. This PB wash is pulled through the QIAprep plate at 300 mTorr for 1 min, then ramped up to 1000 mTorr for 1 min to ensure that wash from the slower wells passes through. Next, the first PE wash is performed by adding 850 μL to each well, pulling through at 300 mTorr, then ramping to 1000 mTorr for 2 min to draw through all wash. For the second of the PE washes 900 μL is added and pulled through at 1000 mTorr for 10 min, followed by transport of the QIAprep plate to the custom drying position (Fig. 3 position 4) set at 80° C where the plate is dried for 2 min.
The plate is then picked up by the gripper and dropped to tap out any residual wash solution. Heated drying is continued for another 2 min. The Lucite top to the QIAvac 96 filtration block is removed to the park position, and the plate holder for waste wash is removed to the wash plate holder position. A clean Matrix 2D bar-coded 1.4-mL deepwell plate is placed to receive eluted plasmid in the lower position of the QIAvac 96 vacuum filtration block. The Lucite top is placed over this plate.
The dried QIAprep plate with bound plasmid is taken by the gripper and placed into the top position over the Matrix receiver plate. The elution is performed by adding either two 75-μL aliquots (mutagenesis run) or two 100-μL aliquots (reproducibility run) of elution buffer (EB). The first EB aliquot is taken up by the eight pipets, added to each of the wells of the QIAprep plate, and allowed to solubilize the plasmid for 1 min. Then vacuum is applied for 5 min at 1000 mTorr to elute the first plasmid aliquot of EB. The vacuum is turned off, and the second aliquot of EB is added to the wells of the QIAprep plate and allowed to incubate for 1 min. The vacuum is then applied strongly and fluttered to ensure complete removal of the eluate. Recovery volume of plasmid averages 126 μL (mutagenesis run) or 160 μL (reproducibility run).
The process is repeated for the second plate, starting from the addition of N3 through elution with PB and PE. Once the process is completed for the second plate, the next two plates of the four-plate process are subjected to the procedures using the same XAP protocol. The entire process takes 374 min including long- and short-term storage for all four 96-well culture block plates.
Yield and Quality of Plasmids Produced on Liquid Handler
For the mutagenesis run, the plasmid concentration in several randomly selected wells containing plasmids produced on the liquid handler was determined with a NanoDrop fiber optic spectrophotometer using 1.5 μL of sample (wells marked “X” in Fig. 6). The average yield per well of a 96-well plate for all plasmids sampled from cultures in LB or TB medium was 3 μg (Table 1, top). There appears to be no notable difference between growth in the different media under the same incubation conditions of 37° C, 250 rpm, for 18 h.
Yield and quality of plasmids prepared on liquid handler, robustness and consistency of plasmid production, and comparison of time required for the automated and manual plasmid production

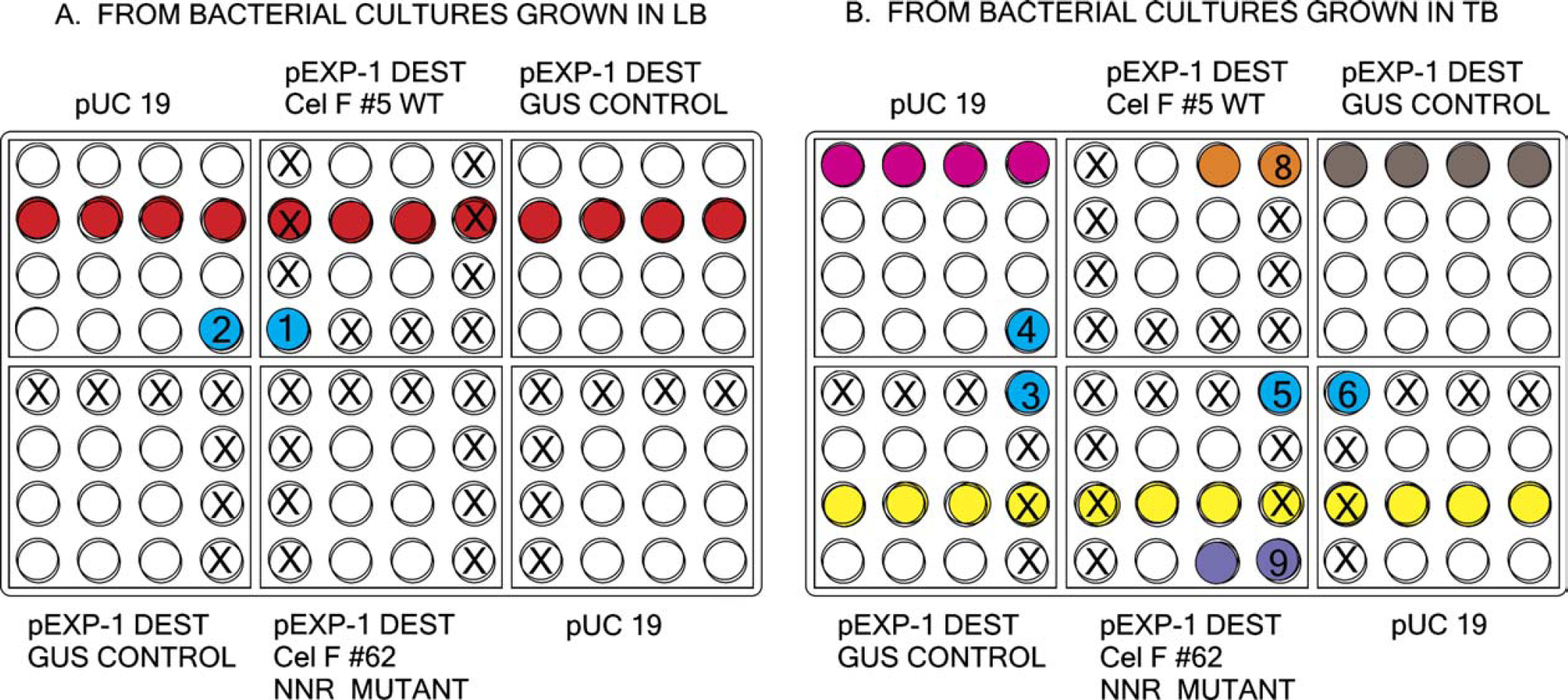
Plasmid preparations obtained from the liquid handler and stored in Matrix deepwell plates from bacterial cultures grown in (A) LB and (B) TB. Samples from (1) wells shown in red and yellow were used for restriction reactions (Fig. 7); (2) numbered wells for Western blot of in vitro proteins (numbers 1-6, 8, and 9 correspond to lanes with those numbers in Fig. 11); and (3) wells in pink, orange, gray, and purple for in vitro azo-CMcellulose plate of in vitro protein (Fig. 12). Wells marked with an “X” were used for concentration determination (Table 1, top).
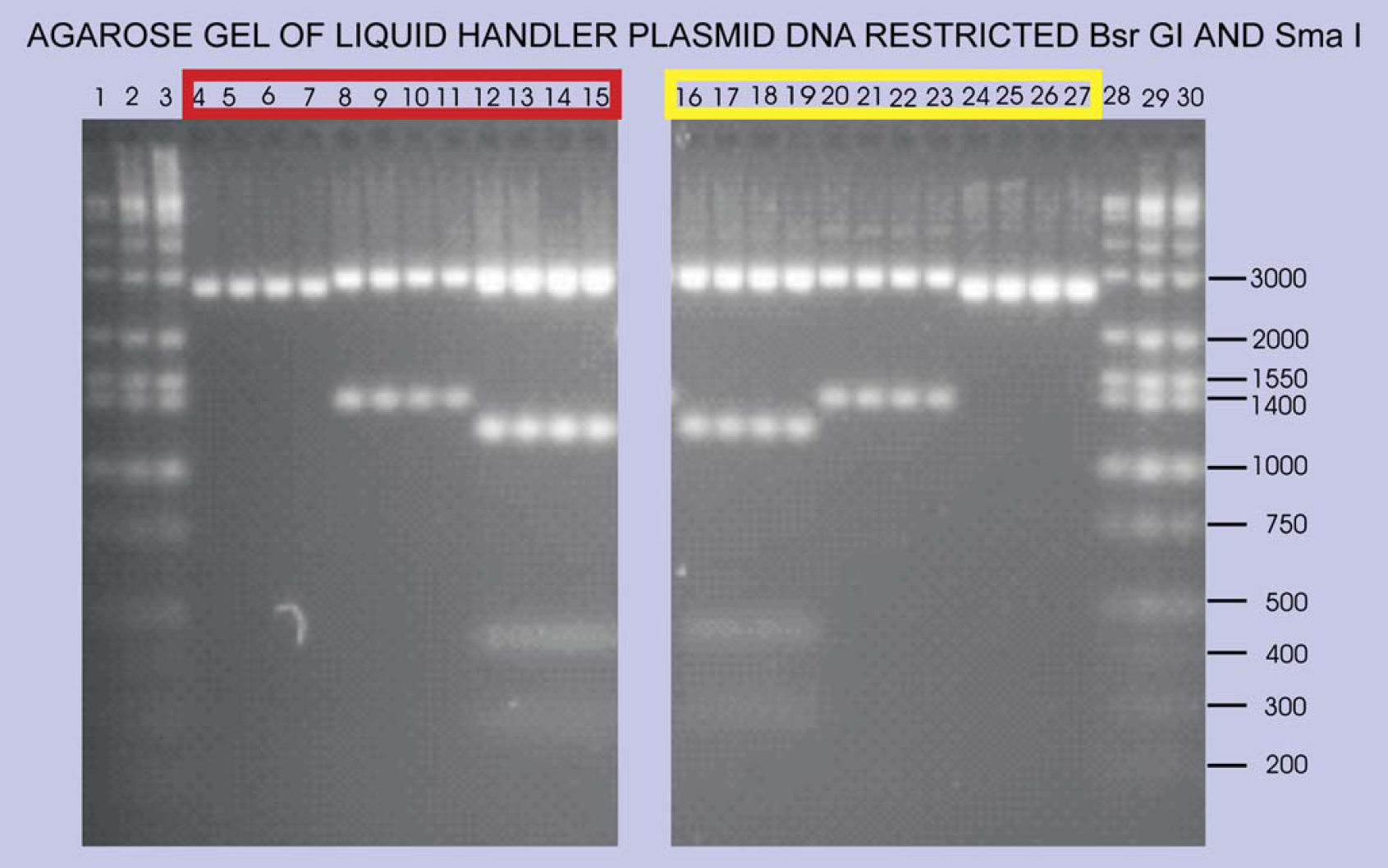
Agarose gel of Sma I and Bsr GI restriction reactions using plasmid DNA samples from the wells shown in red (from LB) and yellow (from TB) in Figure 6. Lanes 1-3 and 28-30: Minnesota Molecular Inc. markers; lanes 4-7 and 24-27: pUC19 cut with Sma I giving band at 2686 bp; lanes 8-11: pEXP-1 DEST CelF #5 giving band at 1364 after Bsr GI cut to release insert; lanes 20-23: pEXP-1 DEST CelF #62 releasing a 1364 bp insert after Bsr GI cut; lanes 12-19: pEXP-1 DEST GUS control vector giving appropriate Sma I and Bsr GI bands at 3113, 1167, 426, and 287 bp (band at 16 bp not visible).
To demonstrate that the plasmid production process on the liquid handler is robust and reproducible, an experiment was designed using one plasmid expressing an active cellulase F mutant (#62) in repeated runs and determining the amount of plasmid in each well. The results of four 96-well plates or 384 wells from the repeated runs are given in Table 1 (bottom). The yield is affected by the amount of bacterial pellet used initially as seen in the automated reproducibility run where incubation time was increased from 18 h to 24 h at 37°C and 250 rpm giving an average yield of 5.35 μg (Table 1, bottom) compared to a 3 μg yield for the 18-h incubation. We are investigating the volume of starting culture and quantity of cells per unit volume that will give optimum yield of plasmid in the shortest run time. For the small amount of culture used here, collections on average were 126 μL for the mutagenesis run using two EB elutions of 75 μL each versus an average recovery volume of 160 μL for the automated reproducibility run using two EB elutions of 100 μL each. The manual run by comparison had a slightly lower average recovery volume of 145 μL using two EB elutions of 100 μL each.
The average yield of plasmid for the automated reproducibility run was 5.35 ± 0.78 μg and for the manual process it was 6.91 ± 0.91 μg (Table 1, bottom) showing that the plasmid production process on the liquid handler is reliable and repeatable, giving a consistently assured amount of plasmid. When comparing the quality of the plasmid from the manual run to that from the automated run we see no difference in the average A260/A280 ratio (both are 1.80; Table 1, bottom). The CelF mutagenesis run had an average A260/A280 ratio of 1.67 ± 0.12, which is not significantly different (Table 1, top).
Table 1 (bottom) also presents a comparison of the total time required by the liquid handler versus that required to process four 96-well plates manually including centrifugation and preparation of long- and short-term storage cards. The liquid handler performed the operations in 374 min whereas it took 441 min to perform manually. Considerable time was saved by eliminating the need for the operator to locate the proper wells for sample delivery manually, which the liquid handler did automatically. This delay manually outweighed the increased washing necessitated by not using disposable tips in the automated process, which saves cost considerably.
Restriction analysis using Sma I and Bsr GI was also performed on the plasmid DNA that was produced on the liquid handler. The plasmids from the LB cultures selected for the restriction analysis are from the row of wells identified by red in Figure 6. The plasmids from the TB cultures were taken from the row of wells identified by yellow in Figure 6. The resulting agarose gel (Fig. 7) suggests that the concentrations determined by the NanoDrop spectro-photometer are correct and that there is no contamination of adjacent wells during the processing of the plasmids by the liquid handler. In addition, examination of sequencing reactions (data not shown) reveals no signs of cross contamination of adjacent wells as indicated by the quality of the chromatograms of the ABI sequence sets (Fig. 8).
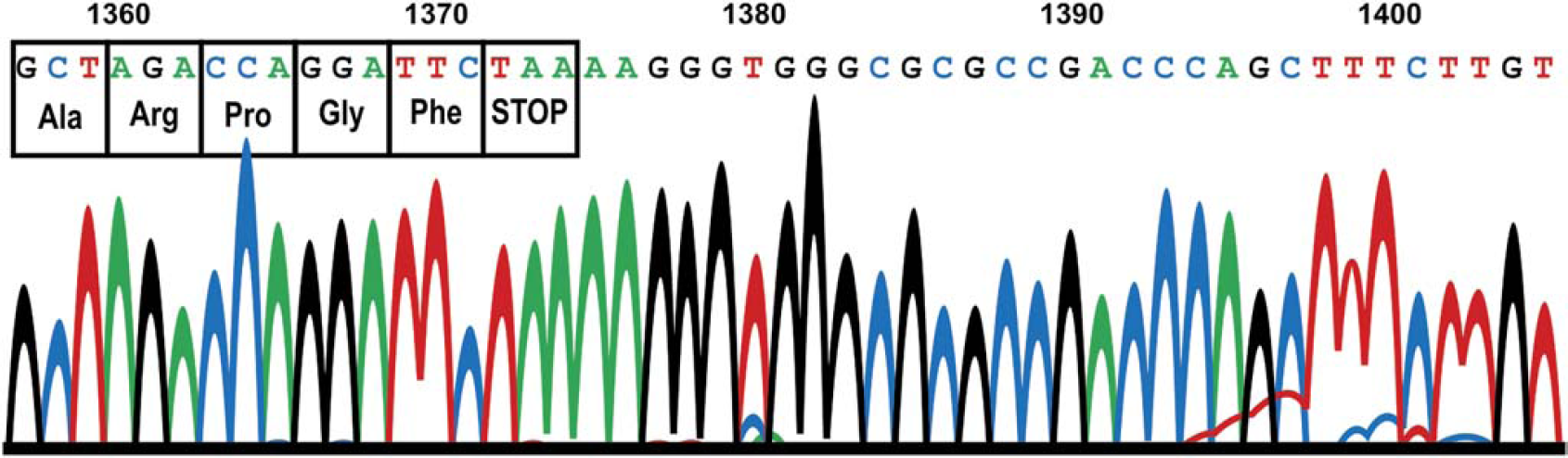
Chromatogram from an ABI 3730 DNA Analyzer for the sequence coding of the last four amino acids at the carboxy end of the pENTR D TOPO 1364 CelF #5 NNR plasmid (from TB cultures) prepared on the liquid handler.
The liquid handler provided enough plasmid to carry out the entire set of workcell protocols (Fig. 4), and the plasmids were of sufficient quality so that all the protocols worked successfully. Results from the completion of these protocols are presented below and show that active CelF proteins are obtained from each of the pDEST expression plasmids produced on the liquid handler.
Plasmid DNA from Gateway Cloning into pDEST 17 and Analysis of Active CelF Proteins from In Vivo Expression in BL21 DE3 Bacteria
The set of pENTR D TOPO CelF NNR library plasmids produced on the liquid handler was LR clonase recombinationally cloned into pDEST 17 plasmid. The pDEST 17 set was transformed into TOP 10 competent bacteria, giving rise to approximately 30 colonies. A transformation efficiency greater than 1 × 106 cfu/μg plasmid DNA was obtained for all plasmid transformations. The pDEST 17 transformants were used in plasmid preparations on the liquid handler to generate plasmids to transform the expression bacteria BL21 DE3. The bacterial cultures were analyzed for expressed protein by chromogenic Western blot using PENTA HIS antibody and showed similar expression levels at 47.7 kDa (Fig. 9 lanes 3-14).
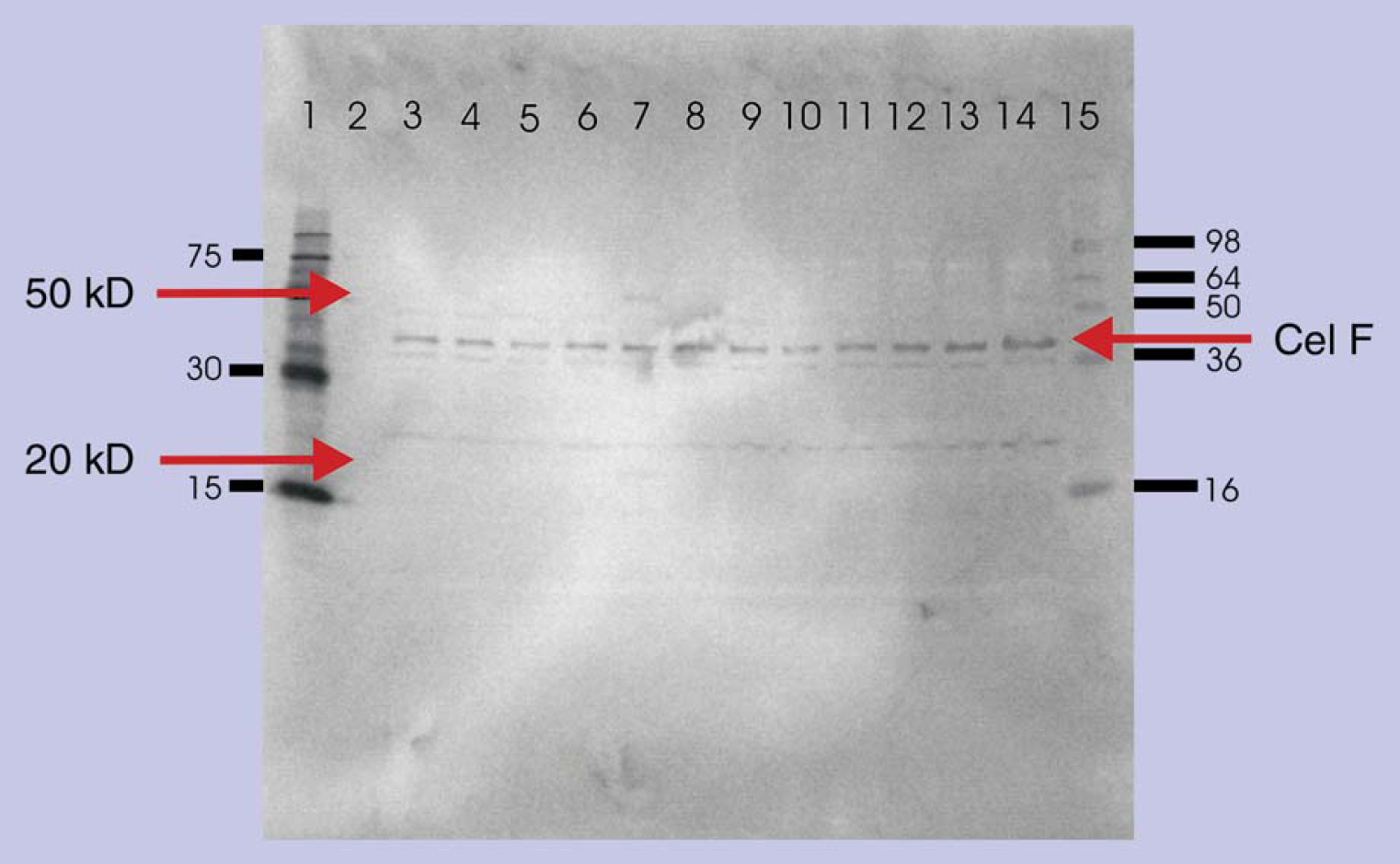
Western blot of proteins expressed in vivo from 12 bacterial BL21 DE3 cells showing uniform expression of the respective CelF NNR mutations produced after IPTG induction. Lane 1: 6× HIS Marker Qiagen; lane 2: blank; lane 3: #83; lane 4: #84; lane 5: #85; lane 6: #86; lane 7: #62; lane 8: #60; lane 9: #25; lane 10: #26; lane 11: #27; lane 12: #34; lane 13: #36; lane 14: #41.
azo-CMcellulose plate analysis indicated that several active CelF NNR mutants (Fig. 10) were produced from this small test pool. Only the CelF #62 NNR mutant had significant cellulase activity at pH 5.8 (optimum pH for wild- type CelF activity) and at the reduced pH of 5 (pH for most commercially useful fermentation processes) as indicated by the cleared zones. It was noted that this mutant had two bands at 25 and 50 kDa associated with it on the Western blot (Fig. 9 lane 7) that were not seen for the other mutants.
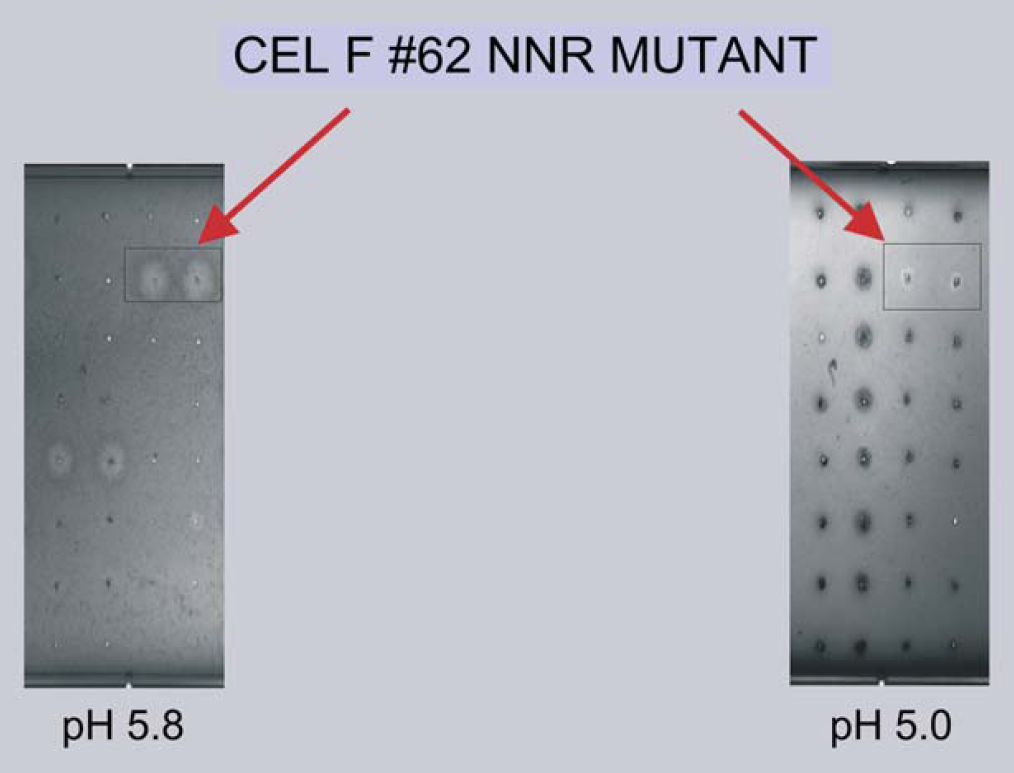
azo-CMcellulose plates spotted with the BL21 DE3 bacterial strain lysates used in the Western gel (Fig. 9).
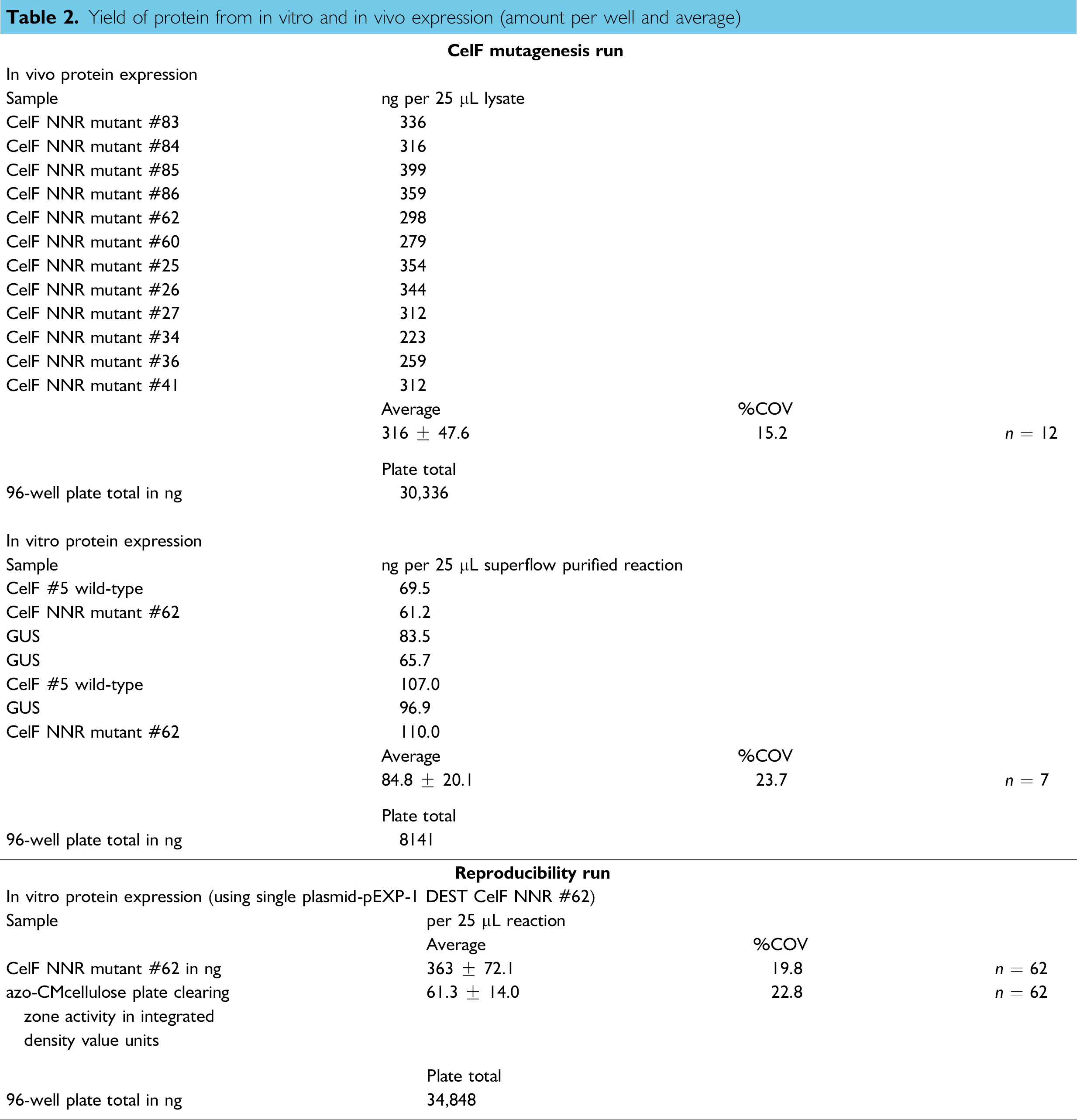
Plasmids prepared on the liquid handler made possible the transformation of BL21 DE3 bacteria, and the resulting production of protein using these cells gave rise to approximately 316 ng protein per well. If all the wells were utilized for the expression of a single protein, a total protein of approximately 30 μg would be obtained (Table 2).
Plasmid DNA from Gateway Cloning into pEXP-1 DEST and Analysis of Active CelF Proteins from In Vitro Expression in Bacterial Lysates Containing T7 Polymerase
The pENTR D TOPO CelF NNR library set containing the CelF #62 mutant clone as well as CelF #5 wild-type clone was also used to generate a pEXP-1 DEST set. This vector provides a modified T7 promoter for in vitro expression from bacterial lysate with T7 polymerase added and generates an amino terminal HIS tag and V5 epitope tag on the CelF ORFs expressed. Plasmid preparations performed on the liquid handler generated pEXP-1 DEST plasmid for in vitro transcription/translation reactions. The protein in the #5 and the #62 samples was purified and adjusted to equivalent concentrations (Fig. 11, lane 8 CelF #5 wild-type and lane 9 CelF #62 NNR mutant). The entire set of in vitro samples for all mutants averaged approximately 85 ng protein per well (Table 2, middle) after purification.
Yield of protein from in vitro and in vivo expression (amount per well and average)
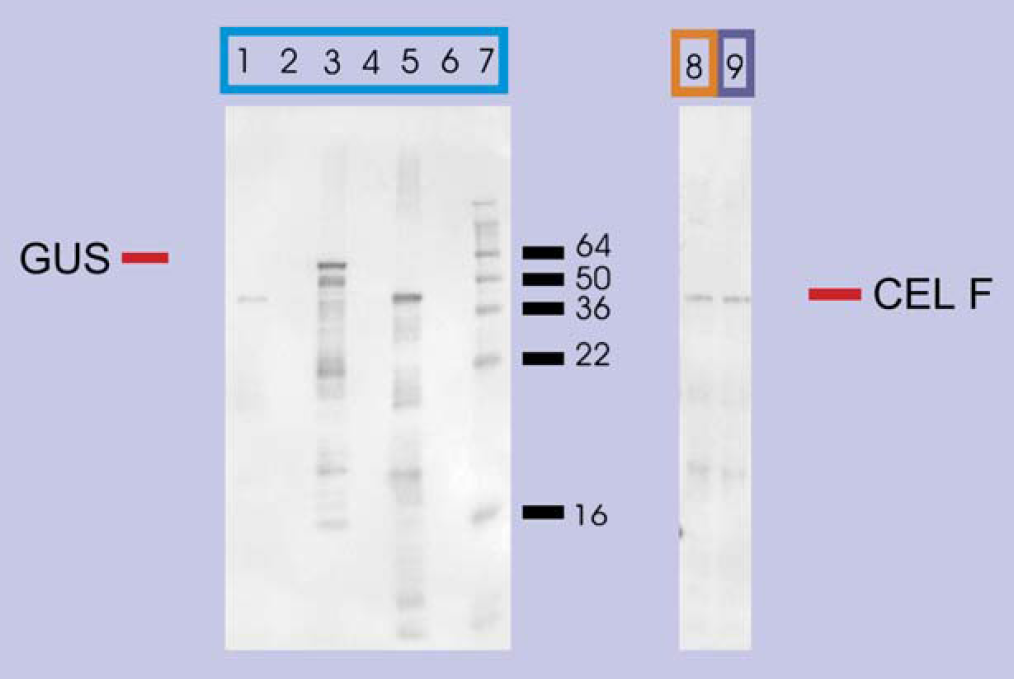
Western blot of proteins expressed in vitro from transcripts generated from pEXP-1 DEST inserts. Lane 3: pEXP-1 DEST GUS expression gives a band at 58.4 kDa; lanes 1 and 8: CelF #5 wild-type gives a band at 49.6 kDa; lanes 5 and 9: CelF #62 NNR mutant also shows a band at 49.6 kDa; lane 7: SeeBlue® Plus 2 Markers from Invitrogen. CelF in lanes 8 and 9 was used in the azo-CMcellulose plate analysis. No expression is observed in pUC19 negative control (lanes 2, 4, and 6).
A sample set of 62 wells was analyzed for CelF #62 mutant protein expressed via in vitro transcription/translation from the pEXP-1 DEST CelF #62 NNR plasmid produced by the liquid handler in the automated reproducibility run. The average yield was 363 ± 72.1 ng per well (%COV 19.8) with an average integrated density value (directly proportional to activity) for the cleared zone of 61.3 ± 14.0 (Table 2, bottom). The quantity and quality of protein produced from the single plasmid preparation in the reproducibility run on the liquid handler demonstrate that the liquid handler process is repeatable and consistent for the expression of functional proteins.
Contamination potential can be assessed by examining results for adjacent wells from in vitro transcription/translation (Fig. 11, lanes 1-6). No sign of GUS (β-glucuronidase expressed by pEXP-1 DEST GUS negative control vector) is detected in any of the other lanes. In addition, CelF is not seen in the GUS or the pUC19 lanes. In vitro transcription/translation is very sensitive and would show distinct signs of contamination if even very small amounts of contaminating plasmids were present.
The activities of the resulting CelF proteins were assayed on an azo-CMcellulose plate at reduced pH 5.0. Further analysis of the #62 clone using expression from the pEXP-1 DEST vector was performed to compare its activity with that of the #5 wild-type CelF ORF (Fig. 12, orange box is CelF #5 wild-type and purple box is CelF #62 NNR mutant). Much greater activity was demonstrated by the CelF #62 mutant than by the CelF #5 wild-type at this pH. This follows the trend of the in vivo data for these clones (Fig. 10 and data not shown). The in vitro protocols can be used for the workcell with the plasmid derived from the liquid handler for a more rapid assay of mutations than the in vivo route.

azo-CMcellulose plate analysis of proteins expressed in vitro (Fig. 11). No cellulase activity is seen for pUC19 or GUS.
Plasmid DNA from Gateway Cloning into pYES2 DEST 52 and Analysis of Active CelF Proteins from In Vivo Expression in INVSc 1 Yeast
An important use of the workcell is to generate yeast strains with enhanced properties by mass transforming the full-length cDNA libraries containing optimized ORFs into yeast. To test the workcell protocols for transforming yeast, a pENTR D TOPO CelF NNR mutant set was cloned into pYES2 DEST 52 and transformed into TOP 10 bacteria. Plasmid preparations of the pYES2 DEST 52 CelF NNR library on the liquid handler were used to transform yeast INVSc 1. Three of the 52 yeast colonies generated were assayed for cellulase activity. Two negative controls (yeast strains expressing spider toxin and frog toxin genes) showed no cellulase activity on azo-CMcellulose plates. The INVSc 1 yeast lysate with the CelF #62 NNR mutant exhibited activity similar to that of the BL21 DE3 bacterial lysate containing that mutant (Fig. 10), with maximum activity at pH 5.8. The enzyme expressed in yeast showed activity at pH 5, 5.8, 6, 7, and 8 but not at pH 4 (Fig. 13). The yeast transformation protocol will be suitable for placement on the workcell using plasmid DNA produced on the liquid handler.

azo-CMcellulose plates at pH 4, 5, 5.8, 7, and 8 spotted with lysate from yeast INVSc 1 expressing CelF #62 NNR mutant. No cellulase activity was seen for the negative controls (wolf spider toxin Lycotoxin 1 and frog toxin Dermaseptin).
Sequence of Active CelF Mutant
The pENTR D TOPO CelF #62 and the pENTR D TOPO CelF #5 plasmids were sequenced. The sequences revealed that the only differences between these clones occurred in the last four amino acids, RPGF for the wild-type clone #5 and PVRE for mutant clone #62 (Fig. 14), suggesting these amino acids might be important for the activity of CelF #62 NNR mutant at lower pH.
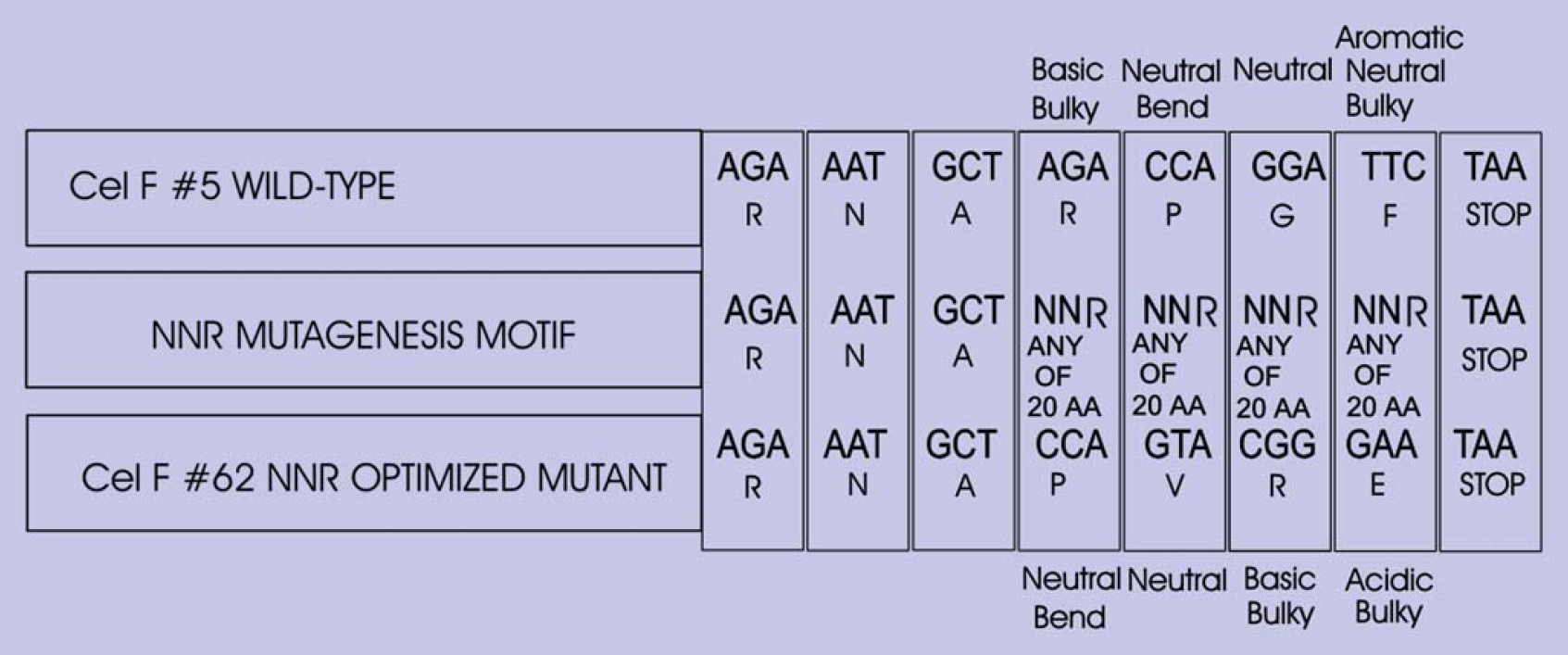
Sequences of CelF #5 wild-type and CelF #62 NNR mutant showing the NNR codon alteration strategy.
Conclusions
A track-based liquid handler designed specifically for 96-well format plasmid preparation was assembled. The liquid handler was incorporated into a plasmid-based functional proteomic robotic workcell and used to provide plasmids to test the protocols for the workcell. The quantity of plasmid produced allowed several parameters to be measured on one set of samples. A small starting culture was used (1.347 mL) and the resulting bacterial lysate was processed. An average volume of 160 μL of plasmid was collected. The average amount of plasmid obtained was 5.35 μg per well for the automated process. There was relatively no difference in the amount of plasmid DNA obtained from LB or TB culture. No signs of contamination between wells were detected. The entire four 96-well plate process on the liquid handler, including long- and short-term storage, pelleting of the incoming bacterial cultures, and execution of all steps in a sterile fashion without disposable pipet tips, which was integrated into the robotic workcell, can deliver plates to the workcell in just under 374 min. All equivalent operations performed manually would take at least 441 min. Plasmid production by the liquid handler was shown to be robust and repeatable, and plasmid quality was equivalent to that obtained manually. The plasmids were used in various expression strategies to produce protein for functional assays to assess protein activity and level of expression in vitro and in vivo. On average, 88 μL of pENTR plasmid set remained and could be stored for future reactions. A liquid handler of this speed and capability is able to perform all protocols necessary for the plasmid-based functional proteomic robotic workcell.
Acknowledgments
We would like to acknowledge the technical assistance and contributions of John Jackson and Jonathan Legazo in BBC NCAUR and Ryan Burdick in FBT NCAUR. We also wish to thank Jody Robinson in the MGB Research Unit, NCAUR, Peoria, IL, core sequencing facility, for her work in obtaining the pUC19 and CelF wild-type and mutant sequences. We would like to express our appreciation to Karen Hughes for her help in formatting and editing this manuscript. We also greatly appreciate the valuable comments and suggestions provided by Dr. Timothy Leathers.