
Abstract
The formation of a molecular aggregate in a confined, nanodimensioned region of space leads to what might be termed a ‘molecular nanoaggregate’. The present review deals with a theoretical formulation termed ‘aggregation-enhanced Raman scattering’ (AERS), and its use in discussion of relative Raman band intensities and selection rules for nanoaggregates. AERs represents a concept for discussion of nanoaggregates that is different from those provided by resonance Raman scattering, surface-enhanced Raman scattering and Mie scattering, all of which ignore the impact of aggregation of molecules on Raman scattering. Beyond the theoretical formulation behind the AERS phenomenon, also outlined in this review are representative samples of the publications of other authors and researchers using AERS to provide explanations for experimental findings. In addition to clarifying issues regarding the use of nanocomposites involving aggregated molecules, it is found that increasing use of AERS concepts is being made to rationalize Raman spectral observations in a range of other disciplines that fall in both the physical sciences and the medical fields.
1. Introduction
The vibrational Raman spectrum of a scattering species that is composed of molecular monomers that are not covalently bonded to one another (but held together by dispersion and electrostatic forces that lead to the formation of an aggregate species) is often found to have vibrational band intensities (associated with ground electronic state vibrations of the monomer) that are enhanced when compared to those of the isolated, non-aggregated monomer. Moreover, the Raman bands observed for the non-covalently coupled species (i.e., the aggregate) are often found to be essentially unshifted spectrally from those of the isolated monomer, with their relative intensities often principally dictated by the wavelength of the exciting radiation. Also, upon resonantly exciting the aggregate's excited state (often referred to as the ‘molecular exciton state’), new Raman bands are evinced that can be associated with lattice motions - typically motions in the aggregate formation direction. Generally, in a molecular aggregate, the active molecules that are coherently engaged in Raman scattering (as well as, for that matter, in fluorescence) occupy a nano-sized spatial dimension. Indeed, in the literature the nanodimensioned length scale defined by the coupled, coherently responding monomers is called the ‘coherence length’. This length is many orders of magnitude smaller than, for example, the size defined by the number of monomers in a physical aggregate, which often has macroscopic spatial dimensions, as observed by flow dynamics and optical polarization measurements.
As a result of their nanodimensioned character and enhanced Raman scattering cross sections, nanoaggregates, especially when they are confined to nano-spatial dimensions, for example, by occlusion within nanoporous materials (forming nanocomposite materials), have several practical applications. For example, used as robust chemo- and bio-sensors, Raman optical imaging chromophores, and as biomarkers.
Often researchers conducting such aforementioned studies (in which the molecular aggregate is the functional component) attempt to exploit Raman scattering, with its intrinsic high resolution structural information content, to gain sensing, positional or reaction pathway information for a process. However, they limit themselves to rationalizing results using concepts such as surface-enhanced Raman scattering (SERS), resonance Raman scattering (RRS), and Mie scattering. Such analyses are inappropriate for systems involving aggregated molecules. The author, however, has advanced a theoretical formulation that aids in interpreting Raman spectra and band intensities for such nanostructural aggregated active elements, and has termed it ‘aggregation-enhanced Raman scattering’ (AERS) [1] (a concept originally published by the author in 1986). In this formulation, molecular exciton theory is used to define both the excited states and wave functions in the Raman scattering quantum formulation.
In the intervening years up to 1986, a limited number of researchers used AERS as a basis for discussing both resonant and non-resonant Raman scattering behaviours by aggregated molecules. Indeed, contributions to the literature dealing specifically with the Raman spectra of aggregated molecules using the aforementioned theory emanated mainly from the author's laboratory. Yet more recently, a number of published works have embraced AERS concepts to rationalize Raman scattering observations for several systems, and have exploited relative Raman intensity measurements for applications in both the physical sciences and medical research fields. In this review, the author revisits the AERS theoretical scheme and briefly surveys the literature regarding publications that use AERS to provide explanations for experimental findings, as reported by the author as well as by a number of other researchers. The present review is not a review of the status of the science dealing with aggregated molecules. Excellent reviews [2,3] and a recent book [4] on the subject are available. A principal aim of this review is to expose more of the scientific community to what has become a crucial conceptual framework in understanding and exploiting Raman scattering by molecules that form molecular aggregates. In particular, in recent years a large number of research publications have dealt with the encapsulation of molecules in single-walled carbon nanotubes (SWNTs), which in many cases leads to the formation of nanoaggregate structures, with the aim of making use of such entities as nanodimensioned tags (i.e., nanoprobe labels) for sensing, optical imaging and following biological reaction dynamics. Often, Raman scattering is a key experimental technique in such studies [5–9]. However, despite results that show the clear finger-print of AERS, researchers seem unaware of the opportunity offered by the concept in aiding their analyses. It is anticipated that this review will provide information that facilitates advances in such areas.
In the present review, Raman scattering by aggregated molecules is discussed in section 2 from a phenomenological perspective so as to set the stage for the theory behind AERS, which is discussed in section 3 in an abbreviated fashion. In section 4, a broad range of published research that explicitly analyses results using AERS is discussed, including both the author's works (subsection 4.1) and works by others recently reported in the literature (subsection 4.2). For the author's works in this area, a progression of approaches occurred in which early studies - at the time - utilized traditional spectroscopic approaches in which a number of molecules with similar chromophoric units were used to aid in assignments of vibrational bands; also, fundamental properties of aggregates, such as visible spectroscopy and optical dynamics, were measured. A few years later, the author's focus shifted somewhat to applications of aggregated molecules resulting from forming composite structures involving substrates with molecules adsorbed onto the exterior surface of nanoparticles or intercalated within the pores of mesoporous materials. For these studies, AERS investigations took something of a backseat to superradiance, and discussions dealt with the exploitation of the dramatic, exceptional properties of the emissions for analysis and device applications. Since superradiance is not a prime concern of the present review, efforts in this vein shall be omitted from the present paper.
More recent efforts involving AERS from the author's laboratory have exploited quantum chemical computational methods to aid in Raman vibrational mode assignments (which have been used to test concepts suggested by the AERS mechanism) as well as for the determination of their structure. Since those papers that make extensive use of quantum calculations (principally DFT calculations) followed something of a common script, the full panoply of such studies is not discussed. Here, only the effort involving the prototypical cyanine dye 1,1′-diethyl-2,2′-quinocyanine (also referred to as ‘PIC’; see Diagram 2) with halide counterion is discussed. For other cyanine dyes, for which quantum chemical calculations have been published, reference information is provided.
In subsection 4.2, a discussion is provided of the increasing number of publications by other researchers that have dealt with application of AERS concepts to explain Raman spectral observations in the physical sciences and medical fields. No attempt is made to be exhaustive regarding the literature - rather, publications that the author believes, through their references and focus areas, provide important information, are briefly described.
Finally, in the Conclusions section (section 5), discussions are provided concerning the growing acceptance of the AERS model and which ongoing research activities represent the centre of gravity for the exploitation of the AERS phenomenon, such as how the relative intensities of various bands can give insight into the alignments of coupled molecules within a structure for cases of non-resonance and resonance excitation.
Recently, research papers dealing with Raman scattering by encapsulated molecules in carbon nanotubes [a–e], which in some cases clearly involve the formation of nanoaggregates, have been published; but the authors appear to be unaware of AERS as a possible framework for understanding their observations. The present review will hopefully add new insight to such publications.
2. Phenomenological Description of the Enhancement Process
Molecules that aggregate in their ground state may result in enhanced polarizability for the resultant structure, which can then couple more strongly to radiation than is the case for monomeric species. Upon excitation, the aggregate can support the formation of molecular excitonic states. These latter states, as with the aggregate's ground state, have a full complement of vibrational states for the various vibrational modes that exist; phonon lattice modes that involve motions of atoms forming the supramolecular structure are also present. For Raman scattering, the existence of excitonic states as well as the coupling between excitonic states affects the aggregate's Raman scattering cross-section because the participation of more energy states for the system allows an enhanced response to incident radiation.
A pictorial representation of the energy-level diagram for Raman scattering by a J-aggregate system (with a band energy lower than the monomer's energy level) is shown in Diagram 1; a fundamental band transition between an initial state i (i.e., v″ = 0) and j (i.e., v' = 1), associated with the difference between the incident and scattered radiation frequencies, is indicated; also, each mode of vibration may contribute a spectral band to the full Raman spectrum. The excited aggregate state is shown to be a band composed of supramolecular levels derived from intermolecular coupling between the N monomers of the aggregate, resulting - in general - in the formation of N levels. In this diagram, s1 represents the excited state of the isolated monomer (which technically does not exist once the monomer is incorporated into the aggregate).
In Diagram 1, which considers AERS, the incident radiation energy is shown to be out of resonance (A) and in resonance (B) with the exciton state; the diagram ignores the existence of phonon modes.

Diagram 1: (A) Non-resonance Raman scattering. The shaded region represents vibro-excitonic bands that are excited when radiation (‘blue’ transition) that does not overlap states of the aggregate impinge on the aggregate system. The ‘green’ transition corresponds to vibronic scattering. Implicit is the assumption that the electron in the excitonic state moves over the coupled molecules in the aggregate, resulting in a spearing effect that results in the vibrations obeying essentially the same Schrödinger equation as for the ground state. (B) Resonance Raman scattering where the incident exciton overlaps a small number of exciton bands. Such an excitation would be expected to result in the further enhancement of vibrational bands. The s1 level corresponds to the monomer absorption band. We found that the exciton of this band results in combination and overtone bands in the Raman spectrum of the aggregate.
3. Theoretical Basis for Aggregation-enhanced Raman Scattering
In a range of research studies, the author and co-workers have shown that the enhancement of Raman bands for aggregated molecules via AERS [1,10–12] (which utilizes Herzberg-Teller intensity borrowing as developed by Albrecht [13]) is mainly attributable to the existence of molecular exciton states and the concomitant enhanced polarizability of the aggregate structure.
Intrinsic to the theoretical formulation of molecular aggregates is the use of excitonic coupling within the so-called ‘J-aggregate’ supramolecular structure derived from non-covalent association between monomeric species. Figure 1 depicts the general situation where non-covalent associations can give rise to both a J-aggregate and/or an H-aggregate, such that, respectively, the aligned monomers within the (simplistic) aggregate structure can assume head-to-tail (top right) or a head-to-head (bottom right) arrangements. The intermolecular forces that lead to the aligned structures between the basal planes of unaligned monomers (left in Figure 1) can arise from π–π interactions as well as Coulombic interactions involving charges on the monomers and counter ions that may be distributed between the monomers. In general, the configurations of monomers in the J- and H-aggregates result in the formation of exciton bands that lie lower or higher, respectively, in energy than the monomer's first excited electronic state. Although the monomers shown in Figure 1 are depicted as being planar (for ease of presentation), real monomers (such as the cyanine dyes that have been the subject of many of our studies) are, in fact, non-planar. However, key structural elements of non-planar monomers are viewed as assuming the relative orientations shown in Figure 1.

The molecular planes of monomers (on the left) self-align to form molecular aggregates (on the right); an arbitrary number of monomers are shown in the aggregate. In the aggregates, the monomers' molecular basal planes can have a range of non-covalent interactions that lead to the alignment of monomers, including π–π van der Waals associations as well as Coulombic interactions. The upper right structure is identified with a J-aggregate; the lower right structure is identified with an H-aggregate; unaligned monomers are represented as distributed structures on the left.
In addition to H- and J-aggregates, in the early literature linear, ribbon and planar structures of aggregated dyes had been proposed. More recently, complex morphologies have been observed through the use of cryo-transmission electron microscopy (cryo-TEM) for some J-aggregates (for example, the formation of three-dimensional tubular aggregate bundles for the J-aggregate formed by pseudoisocyanine (PIC) [2,14]). All the observed morphologies confirm the importance of aligned monomers in defining the intramolecular interactions in the J-aggregate. Such alignment is implicit in the present formulation through the interaction potential that appears in the quantum mechanical Hamiltonian. As a result, a general point that might be made is that using the wave functions and energies for what might be viewed as a simplified molecular exciton system in the Raman scattering formulation provides a conceptual framework for understanding how the movement of energy among coherently coupled molecules might be expected to affect Raman band intensities and scattering frequencies, even if the structure and couplings that define aggregate formation are more complicated. In other words, the Raman scattering problem to be solved might be viewed as transcending the actual physics of exciton motion among coupled molecules, and to be somewhat akin to solving such ideal problems as the particle-in-a-box, the harmonic oscillator and the rigid rotor, where the actual potential functions used to define such problems are approximations (or simplifications) of more complex interactions that apply for real systems.
An abridged description of the theory is presented below, the full description appearing in the original publication [1].
3.1 The molecular exciton solution
The theoretical approach begins with the quantum mechanical Hamiltonian that describes the internal system of N molecules that can mutually interact through a potential term V, as shown in eq. 1 below. Here,

the interaction potential between molecules m and n, where m is any of the other molecules in the aggregate besides molecule n; i.e., m can have the values 1 to N with the exception of n.
The vibronic wave function for an individual molecule n is given by:

where, implicitly, n labels the position of the molecule and i is a composite quantum number that specifies the number of vibrational quanta of excitation with a known distribution among the vibrational modes. Eq. 2 is the crude adiabatic Born-Oppenheimer approximation, where q is a composite spatial coordinate of the electrons, Q represents the normal coordinates, and Q0 is the normal coordinate for the ground-state equilibrium configuration. Also, φ is a many-electron wave function that depends on the coordinates and spins of the π-electrons, while χ is the vibrational wave function which, more explicitly, in the zeroth approximation and is defined as:

In this latter expression, the subscripts n and ik specify the molecule and the number of vibrational quanta i in mode k, respectively, and Qk is the normal-coordinate variable.
The aggregate ground state is expressed as an antisymmetrized product wave function [15–17] that is defined as:

in which S is the number of π-electrons,
The excitonic state is developed from the premise that only one molecule of the aggregate is excited and that a superposition of spatially separated molecular wave functions must be taken, since quantum mechanically we are unable to specify which molecule is excited. A particular arrangement might be identified as:

where φn i is an excited vibronic state of molecule n and i is the composite vibrational quantum number identified above. The wave function Φni must also be antisymmetrized, leading to the antisymmetrized wave function θni defined below:
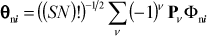
where the notation is identical with that used in eq. 4. The stationary-state wave function is found from eq. 6 by allowing for excitation at any molecule n. The resultant equation is:
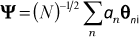
where an is related to the probability that molecule n is excited.
Upon defining the secular equation for the coefficients an in eq. 7, we have:

where the prime indicates that n ≠ m. This equation represents a matrix with diagonal elements -anε and immediate off-diagonal elements an±1 M, with M = Vnn±1 (i.e., the matrix element of the interaction potential for state Φni in eq. 5).
As a result, the allowed energies (for the so-called ‘J-aggregate’) are:

which, after substitution in eq. 9 and the determination of the expansion coefficients, allows - with clever manipulation [1] - the normalized vibro-exciton wave function to be expressed as:
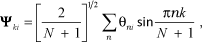
where k is the electronic quantum number label, which can have integer values of 1 to N. The sum over n (the molecule label) also goes from 1 to N.
Eqs. 9 and 10 were then used in Raman scattering theory as excited-state energies (referenced to the first excited level of the isolated molecule) and wave functions, respectively.
3.2 Raman scattering between the ground and vibro-excitonic states
The Raman intensity in a particular direction Κ′ (for an incident radiation vector κ) can be defined as shown in the equation below:

In this expression, Ñ is the number of scattering centres (i.e., the number of aggregates), the sum is over the excited electronic states r, the μs are transition dipole moment vectors, the es are microscopic electric field strength vectors, and Er0 is the energy difference between the upper state r and the ground state 0.
Expressed in terms of polarizability, the Raman intensity becomes:

where i, j, k and l are laboratory coordinates.
At this point, we utilized the Born-Oppenheimer approximation to describe the states of the molecular system. Also, for ease of presentation, we converted transition dipole expressions to Dirac notation, and concerned ourselves with vibrational Raman scattering, where the molecule after scattering radiation remains in its ground electronic state while the vibrational state changes from its initial value v″ to its final value v'.
In [1] it was shown that the polarizability of the aggregate can be written as:
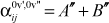
where:
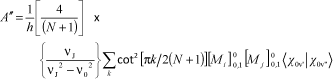
and:

where k is an odd integer ranging from 1 to N, hα1,s is the coupling term between electronic states 1 and s for the molecule with an equilibrium ground-state configuration, Qα is the displacement of mode α, δE01,s = E01 - E0s is the electronic energy difference (again, for the molecule with an equilibrium ground state configuration) and the prime on the summation over s excludes state r from the sum.
Upon summing over the cotangent function, one finds the following:

We also note that in the harmonic oscillator approximation, the A″ term (hereafter referred to as the ‘A term’) is responsible for Rayleigh scattering while the B″ term (hereafter referred to as the ‘B term’) gives rise to the scattering of the fundamentals of the different modes.
We thus determine that the B term acquires a factor of magnitude N resulting from substituting eq. 16 into eq. 15, which reflects the summation over exciton states. Upon squaring, as required by eq. 12 to calculate the Raman intensity, one gets the factor N2 Ñ times other terms which are general in the Raman intensity expression. Ñ, as mentioned earlier, is the number of scattering centres (i.e., aggregates) and, hence, N Ñ represents the number of molecules that scatter radiation. The additional factor of N thus represents an inherent enhancement factor due to the formation of an aggregate containing N molecules.
As with the B term, the A term explicitly involves an additivity factor N (the number of molecules participating in the coherent process) and - additionally -energy differences relative to the excitation frequency that lead to absorption resonances, as well as a factor consisting of sums over excited vibronic states of overlapping integral products, such as
However, even though the excitation conditions may correspond to a resonance situation, the B term may also be responsible for the presence of vibrational bands in the Raman spectrum. For example, for certain bands, contributions from the A term are expected to vanish. This latter situation occurs for the ‘strong-coupling case’ for exciton formation (as detailed by Kasha [21,22]) that corresponds to a slight impediment for the excitation leaving the isolated single molecule and ‘roaming’ through the aggregate structure. In such a case, the Born-Oppenheimer separability of intramolecular electronic and vibrational motions is necessitated, and since the excitation would be spread over many molecules, each individual molecule would have essentially the same electronic structure as a ground-state molecule. Thus, it would follow from the Franck–Condon principle that the A term would only be nonzero for upper state modes identical to ground-state modes. In other words, the A term would not contribute to the appearance of Raman bands resulting from the overlap of intramolecular ground and vibro-excitonic modes. On the other hand, the excited-state lattice modes of the aggregate (resulting from the intermolecular potential function) are expected to give rise to nonzero overlap integral products with ground-state intramolecular modes. Consequently, since we find for systems of aggregated molecules that the same B term bands appear in both the resonance and non-resonance Raman spectra, we can conclude that all the B term modes are most likely fundamental since, with non-resonant excitation, these same modes (as a result of closure over excited-sate modes) must be fundamentals in the harmonic oscillator approximation [23–28].
The B term, in addition to the situation discussed above, can become dominant under two other conditions. First, B alone contributes to Raman bands in the non-resonant Raman case, where vibrational closure over the excited-state vibrational modes (as mentioned above) relegates the A term to make a contribution only to Rayleigh scattering. Second, for non-totally symmetric modes, for which the A term vanishes through symmetry, the B term contributes in both the resonance and non-resonance cases.
It is to be noted that intensity-borrowing through the Herzberg–Teller coupling term (specifically,
A novel approach to deciphering the alignment of monomers within an aggregate has been identified through our studies [12]. This approach involves exploiting non-resonant radiation to assess which motions are intrinsically enhanced simply as a result of the monomers being coupled through the dipolar forces that hold the aggregate together, as well as the determination of which motions are in the aggregate formation direction through the acquisition of resonance Raman spectra of the aggregate. The combination of such spectra gives orientational information with vibrational resolution.
Another point to be made here is that the inherent enhancement factor (N) is akin to the
4. The polymethine dyes and porphyrins investigated
Raman spectral characteristics - as explained by AERS -have been observed in this laboratory for a number of cyanine dyes. Also, molecular aggregation has been found to occur for homogeneous as well as heterogeneous systems, where in the latter the molecules are adsorbed onto substrates. Some of the cyanine dye molecules examined include: 1,1′-diethyl-2,2′-quinocyanine, the prototypical cyanine dye, also referred to as ‘pseudoisocyanine’ (PIC) [7]; 3,3′-dimethyl-9-phenyl-4,5:4′,5′-dinaphthothiacarbocyanine (NTC) [29]; 1,1′-diethyl-3,3′-bis(3-sulfopropyl)-5,5′,6,6′-tetrachlorobenzimidazolocarbocyanine (BIC, also known as ‘TDBC-3’) [30]; 1,1′−3,3′-tetraethyl-5,5′,6,6′-tetrachlorobenzimidazolocarbocyanine (TTBC) [31]; and 3,3′-diethyl-5,5′-dichloro-9-phenylthia-carbocyanine (DDPT) [32]. The basic chemical structures of the cyanine dyes above are shown in Diagram 2 below. As mentioned above, the environments in which aggregation occurs for the aforementioned molecules utilized by the author include homogeneous solutions and adsorption onto surfaces, including in the pores of mesoporous materials. Moreover, we have found a number of porphyrin derivatives as well as some random molecules that also evince AERS. In particular, we have investigated a number of tetraaryl porphyrins: for example, tetraphenylporphyrin (H2TPP), tetrapyridylporphyrin (H2TPyP), tetrakis (p-4-methyl-pyridyl) porphyrin (H2TMPyP4+) and tetrakis(p-carb-oxyphenyl)porphyrin (H2TCPP4-) (see chemical structures in Diagram 3 [33–36]). In all of the reported studies below, measurements were made at room temperature.

Cyanine dyes used in aggregation studies

Tetraaryl-substituted porphyrins used in aggregation studies
4.1 Discussion of the selected studies involving AERS from the author's laboratory
4.1.1 D. L. Akins, C. Akpabli and X. Li, “Surface Potential Dependence of Enhanced Raman Bands of Aggregated Cyanine Dyes,” J. Phys. Chem., 93 , 1977–1984 (1989). [11]
In this paper, the surface potential dependence of Raman scattering by aggregated molecules was analysed using molecular vibro-excitonic states as excited states in the scattering problem. It was shown that when the incident excitation frequency was close to resonance with the J-aggregate absorption, Raman bands attributable to the A term of Albrecht's polarizability expression appear, along with bands attributed to the B term that are also present in the non-resonance case (see Figures 2 and 3). The former bands were interpreted as due to the Franck-Condon overlap of the intramolecular modes of the molecules in the aggregate with the intermolecular lattice modes of the aggregate. It was deduced that bands assigned to the A term can be used to normalize the intensity of bands due to the B term as regards both the excitation frequency and surface concentration, thus serving as internal Raman standards and thereby isolating the electric potential dependence of Raman bands assigned to the B term. In this study, we determined that a Stark-effect shifting the J-aggregate band can explain the voltage dependence of the excitation frequency and concentration normalized Raman band intensities for aggregated 2,2′-cyanine and 4,4′-cyanine.
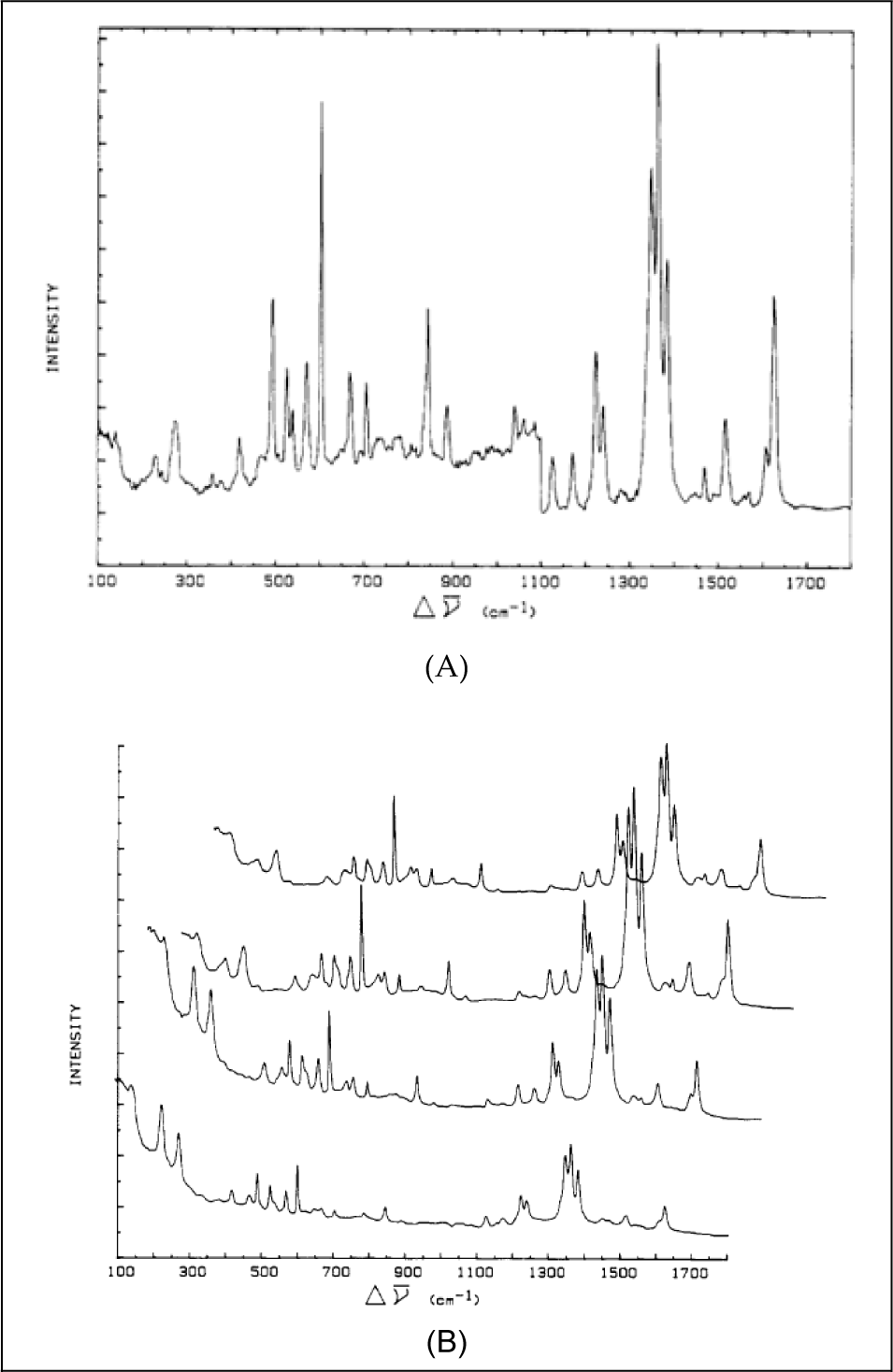
Raman spectra of aggregated 2,2′-cyanine (dye (1) in Diagram 2) excited by non-resonant (see panel (A)) and resonant radiation (see panel (B)). Highly polished Ag electrodes were used. Panel (A) shows the spectrum of 5 × 10−7 M 2,2′-cyanine chloride excited by 488-nm argon ion laser radiation. The spectrum below 1,100 cm−1 has a full-scale setting of 4 × 103 counts/s, while higher wavenumber bands have a scale setting of 1 × 103 counts/s. Laser power was ca. 30 mW for the sample. Panel (B) shows the spectrum of 5 × 10−7 M 2,2′-cyanine chloride, excited by 583-nm laser radiation from an argon ion pumped, rhodamine 6G dye laser. The voltage-dependent spectra, for potentials −0.8, −0.95, −1.10 and −1.2 V all vs. SCE, are provided from bottom to top, respectively. The scale setting in all cases is 3 × 104 counts/s and the laser power for the sample was ca. 30 mW.

Raman spectra of 4,4′-cyanine adsorbed onto a polished silver electrode at −0.6 V vs. SCE from a 5 × 10−6 M, 0.1 M KC1 solution at pH = 12, and an excitation wavelength (A) of 514 nm and (B) of 647.1 nm. The solution was allowed to equilibrate with the electrode for at least 2 h before spectra were taken. (Taken from [37], Figure 2).
4.1.2 D. L. Akins, J. W. Macklin, and H.-R. Zhu, “Fourier Transform Raman Spectra and the Structure of Adsorbed 2,2′-Cyanine,” J. Phys. Chem., 95 , 793–798 (1991). [38]
Near-infrared excited FT-Raman spectra of 2,2′-cyanine (thus avoiding resonance excitation effects) adsorbed onto a smooth silver electrode and of polycrystalline 2,2′-cyanine (see Figure 4) were compared with dispersive Raman spectra obtained using visible excitation to determine the adsorbate structural characteristics. The Raman spectra were interpreted as indicating that 2,2′-cyanine exists as a structural composite of polycrystalline and two intermolecular arrangements of cyanine monomers forming the J-aggregate. This structural model was shown to explain Raman intensity variations that depend upon dye concentration, supporting electrolyte, pH and electrode potential. Variations of the FT-Raman relative band intensities over a surface potential range in which the surface concentration was increased indicated that one of the structural conformers in the J-aggregate is thermodynamically favoured at more negative potentials. Finally, the structural model and proposed makeup of the adsorbate were used to rationalize the effects of electrode potential on relative band intensities in FT Raman spectra.

Near-infrared excited FT Raman spectra of 2,2′-cyanine. Full-scale relative intensities are 300:1:1 for (A), (B) and (C), respectively. (A) FT-Raman spectrum of a polycrystalline dye in a melting point capillary. Instrument resolution was set at 1 cm−1, laser power was 1 w, FT scan range was set from 10 to 3500 cm−I, and 400 scans were accumulated (taking ca. 13 min). (B) Raman spectrum of dye adsorbed onto a smooth silver electrode from a solution of 5 × 10−4 M dye, 0.1 M KCI and 0.01 M NaOH, at an electrode potential of −0.6 V vs. SCE. Spectrum was excited by using 1 W of 1.064-nm radiation, had a nominal resolution of 4 cm−1 and was acquired in 400 scans. (C) FT-Raman spectrum of dye adsorbed onto a smooth silver electrode from a solution of 5 × 10−4 M dye, 0.1 M KCI and 1 M HCI, at an electrode potential of −0.8 V vs. SCE. Spectrum was excited by using 1 W of 1.064-nm radiation, had a nominal resolution of 4 cm−1 and was acquired in 400 scans.
Research from this laboratory has focused on molecular excitons associated with aggregate structures and electron transfer dynamical processes involving aggregated species as one of the reactants. Our general interest derives from: (a) the fact that adsorbed aggregated structures, as formed by spectral sensitizer-dyes (cyanine dye) [39], often find utility in sensitizing semiconductors to light that is not intrinsically absorbed [40]; (b) the ubiquity of aggregated structures in biological systems where they function to convert optical radiation into chemical energy and promote charge-transfer processes [41]; and (c) the prospect that aggregated molecules may find utility as structures with enhanced nonlinear optical susceptibilities [42–45].
4.1.3 D. L. Akins, H.-R. Zhu, and C. Guo, “Absorption and Raman Scattering by Aggregated meso-Tetrakis(p-sulfonatophenyl) porphine,” J. Phys. Chem., 98 , 3612–3618 (1994). [33]
In the subject paper, we combined absorption and Raman scattering measurements to characterize aggregated TSPP (dye-7 in Diagram 3) in an aqueous solution. We discussed the implications of the theory of aggregation enhancement when applied to homogeneous aqueous phase aggregates of TSPP. We have found, as also reported by others [46,47], that homogeneous aqueous-phase TSPP aggregates give rise to a sharp, intense absorption band at 490 nm and a broad, weak absorption band at 422 nm (see Figure 5); this aggregation evolves through the formation of protonated diacid TSPP, and only occurs at a pH below 5; moreover, the TSPP diacid is the repeating unit in the aggregate. We further deduced that the vibrations of adjacent molecules are perturbed in a fashion that suggests that the structure of the aggregate has arrangements that may be characterized as J- and H-types, which are designations commonly used for aggregates of the cyanine dyes [21]. Additionally, vibrational band assignments were made. The variation of the Raman spectrum of the homogeneous system as a function of pH is shown in Figure 6.

Absorption spectra of TSPP in aqueous solutions of pH = 1.6, with a path length of 1 mm: (A) 0.5 mM, (B) 1.0 mM, (C) 2.5 mM and (D) 5.0 mM

Resonance excitation of the Raman spectra of TSPP in an aqueous solution for low- and high-pH ranges. (A) TSPP concentration of 5.0 × 10−5 M, pH = 1.6, λex = 488. (B) TSPP concentration of 5.0 × 10−5 M, pH = 1.6, λex = 514.4 nm. (C) TSPP concentration of 1.0 × 10−5 M, pH = 12.3, λex = 488 nm. (D) TSPP concentration of 1.0 × 10−5 M, pH = 12.3, λex = 514.5 nm. Each spectrum has been normalized with respect to the band at 1,231 cm−1.
4.1.4 Daniel L. Akins, Serdar Özçelik, Han-Ru Zhu, and Chu Guo, “Fluorescence Decay Kinetics and Structure of Aggregated Tetrakis(p-Sulfonatophenyl)Porphyrin,” J. Phys. Chem., 100, 14390–14396 (1996). [37]
In this study, we utilized fluorescence decay lifetime measurements (which provide information on exciton emission dynamics) and resonance Raman spectral data (which provide additional insights into the structure of porphyrin aggregates). Both phase modulation and streak camera fluorescence lifetime measurement techniques were used. We primarily focused on the N-protonated TSPP aggregate.
Typical absorption spectra for the monomeric dianion, the aggregate and the free-base are shown in Figure 7, the presence of a particular species being determined by the pH at which the spectrum was attained.

Absorption spectra of a free-base (peak at 410 nm), a monomeric dianion (peak at 432 nm) and an aggregate (peak at 489 nm) TSPP. The concentration of TSPP in each case is 5 × 10−5 M in the aqueous solution. For the free-base, pH = 12; for the monomeric dianion, pH = 4.5; for the aggregate, pH = 1.6 and [KCl] is 0.1 M. Spectra above ca. 500 nm have been offset by +0.2 absorbance units and amplified by the indicated factor to enhance presentation. The structure on the band near 500 nm for the free-base is an artefact of the absorption spectrometer.
From the emission dynamics, we deduced that an internal conversion competes with the exciton emission process (S2 → S0), the internal conversion being between the S2 (B; or Soret) and the S1 (Q) excited states of the porphyrin. We also ascertained that the fluorescence lifetime of the aggregate depends on whether the electronic excitation is to the B- or the Q-state. Specifically, our phase-modulation results for the emission spectra (see Figure 8) indicate that the lifetime of the free-base is ca. 9.4 ns and that that of the monomeric dianion is ca. 3.5 ns, independent of whether B-band or Q-band excitation was used. For the aggregate, we measured a fluorescence lifetime of ca. 290 ps when a B-band excitation of 488 nm was used, but a lifetime of ca. 82 ps when a Q-band excitation of ca. 706 nm was used. The fluorescence lifetime measurements accomplished with a streak camera were consistent with those found using our phase-modulation instrumentation (any difference between the two being explained as being due to the difference in detection frequency band-pass). Our speculation of the excitation wavelength-dependence of the fluorescence lifetime was that B-band excitation and Q-band excitation result in different coherence lengths for the Q-state of the aggregate from which emission occurs. The indirect B-band excitation of the aggregate requires internal conversion (IC) between the S2 and S1 aggregate manifolds, which implicitly indicates that vibronic states are activated (which are known to cause localization of excitonic energy), resulting in a decreased coherence length. The different coherence lengths (i.e., different numbers of molecules that are cooperatively coupled) translate into different radiative lifetimes, since molecular exciton theory indicates that radiative lifetime is inversely related to the number of molecules in the aggregate [44]. As a result, the radiative lifetime resulting from the indirect excitation into the Q-state would be longer (the 290 ps measurement) than that associated with the direct Q-state excitation.

Fluorescence spectra of free-base (A), monomeric dianion (B) and aggregate (C) TSPP solutions. Respective excitation wavelengths are indicated. The concentration of TSPP in each case is 5 × 10−5 M in the aqueous solution. For the free-base, pH = 12; for the monomeric dianion, pH = 4.5; for the aggregate, pH = 1.6 and [KCl] is 0.1 M. The annotation ×50 indicates the amplification factor used.
We have also been able to deduce, through Raman studies (see Figure 9), very specific structural changes that appear to occur within the porphyrin upon N-protonation and incorporation within the aggregate. We have confirmed some of our earlier determinations, such as the planar conformation of the intercalated monomers, but go further in assigning vibrational frequencies to specific bonds of the porphinato macrocycle and comparing the bond lengths in the aggregate environment to those of the monomeric dianion.

Resonance Raman spectrum of TSPP, where the solution is the same as in Figure 8 and spectra for wavenumbers greater than 400 nm have been amplified by a factor of 5 and offset to aid in visualization: (A) free-base, λex = 416 nm, offset of 10 K counts; (B) protonated dianion, λex = 432 nm, offset of 20 K counts; (C) aggregated dianion, λex = 488 nm, offset of 35 K counts
4.1.5 Chu Guo, Metin Aydin, Han-Ru Zhu, and Daniel L. Akins, “Density Functional Theory Used in Structure Determinations and Raman Band Assignments for Pseudoisocyanine and Its Aggregate,” J. Phys. Chem. B, 106 , 5447–5454 (2002). [12]
For cyanine dye-1 (i.e., (PIC)), we performed quantum chemical calculations for the optimized structure and determined Raman vibrational frequencies based on density functional theory at the B3LYP level using the 6–31G(dp) basis set. We ascertained that the equilibrium structure of ground-state PIC has near-C2 symmetry, with a 46° twist between the planes of the two quinoline moieties that are positioned about the central methine carbon. Vibrational mode analysis of the calculated Raman spectrum suggests that many of the experimentally observed Raman bands between 500 and 1,800 cm−1 for monomeric PIC are associated with totally symmetric in-plane deformations of phenyl and/or pyridyl rings, while several weak bands below 500 cm−1 are attributed to out-of-plane doming and ruffling of quinoline macrocycles. We further noted that: (1) upon comparison of the experimental Raman spectrum of the PIC monomer with the Raman spectrum measured for the aggregated PIC, under non-resonant conditions changes revealed that the aggregation results in enhanced scattering for specific vibrational modes that contain principal contributions from the in-plane deformation of the phenyl ring in the quinolone moiety; (2) with resonant excitation of the aggregate, observed vibrational modes associated with out-of-plane distortions of the quinoline macrocycle were found to exhibit even greater enhancement. The analysis of our Raman measurements for the monomeric and aggregated PICs provides details about the structure of the molecular aggregate. Additionally, the calculation of charge distribution -utilizing the Mulliken population analysis approach -indicates that positive and negative charges are alternately and symmetrically distributed over a conjugated ring system, and that the positive charges among the peripheral hydrogens and those in the N-ethyl side chains are asymmetrically distributed.
Similar studies to that above were conducted for DDPT (ref. [48]) and TTBC (ref. [49]).
4.1.6 (a) W. Xu, H. Guo and D. L. Akins, “Aggregation of Tetrakis(p-sulfonatophenyl)porphyrin within Modified Mesoporous MCM-41,” J. Phys. Chem. B, 105 , 1543–1546 (2001) [17]; (b) W. Xu, H. Guo and D. L. Akins, “Aggregation and Exciton Emission of a Cyanine Dye Encapsulated within Mesoporous MCM-41,” J. Phys. Chem. B, 105 , 7686–7689 (2001) [16]; (c) W. Xu and D. L. Akins, “Absorption and Exciton Emission by an Aggregated Cyanine Dye Occluded within Mesoporous SBA-15,” J. Phys. Chem. B, 106 , 1991–1994 (2002). [50]
Other studies from this laboratory have been reported regarding techniques for forming aggregated molecules within mesoporous aluminosilicate structures [16,17,50]. In one study, we encapsulated tetrakis (p-sulfonatophenyl)-porphyrin (i.e., TSPP) within aluminium-incorporated MCM-41, and provided spectroscopic evidence that confirmed the encapsulation - in particular, the dramatic changes in absorption and emission spectra that occur upon the formation of the occluded aggregate [17]. However, this system has reduced the prospect for the exploitation of the composite material for applications of new devices and materials because the exciton emission (which is indicative of aggregate formation) was weak. The weakness of the emission was due to it coming from the Q-state, as a result of internal conversion from the initially excited B-state of the porphyrin and the low emission cross-section between the Q-state and the ground state.
In a second study, we encapsulated monomeric and aggregated TTBC within MCM-41 and found strong excitonic emission from the aggregated composite [16]. However, for this latter case, the amount of aggregated TTBC formed within the channels of MCM-41 as a result of the synthetic approach used did not dominate the emission from the sample, since a substantial amount of the monomer remained within the mesoporous channels. Moreover, from XRD studies we ascertained that the structural integrity of the aluminosilicate was somewhat compromised upon forming the occluded aggregate. Consequently, this sample also exhibited diminished promise as regards applications.
In another study, we replaced MCM-41 with SBA-15 [50], which has a thicker wall structure and which can be varied within the range 31 to 64 Å, compared to the 10 to 15 Å wall thickness range commonly found for MCM-41. UV-vis absorption spectra for TDBC in three different environments are shown in Figure 10. The composite consisting of J-aggregated TDBC and the modified SBA-15 was found to represent a new, superradiant, robust nanomaterial whose spectroscopic properties emanate from quantum confinement over several coherently responding molecules associated with the coherence length, and whose structural form is further dictated by the restricted growth region defined by the one-dimensional nanoporous structure of the silicate. The emission spectra from TDBC in three different environments are shown in Figure 11.

UV-vis absorption spectra of TDBC in various environments. (A) Homogeneous solution-phase monomeric TDBC at 5 × 10−5 M in a methanol solvent (dashed line). (B) Solution-phase aggregated TDBC, formed from a monomer concentration of 5 × 10−5 M at pH = 11 in an aqueous solvent (dotted line). (C) Aggregated TDBC encapsulated in SBA-15 that was modified by surface silylation using (aminopropyl)-triethoxysilane, designation TDBC-A/SBA-15 (solid line). F(R), the right-hand-side label, refers to the so-called ‘remission function’ (a linear function of the concentration of homogeneous absorbers) for diffuse reflection measurements and is calculated from F(R) = (1 - R)2/2R. In this expression, R is the diffuse reflectance, given by R = J0/I0, where I0 is the incident intensity at the surface and J0 is the intensity of the reflected light [see, for example, Kubelka, P. J. Opt. Soc. Am.
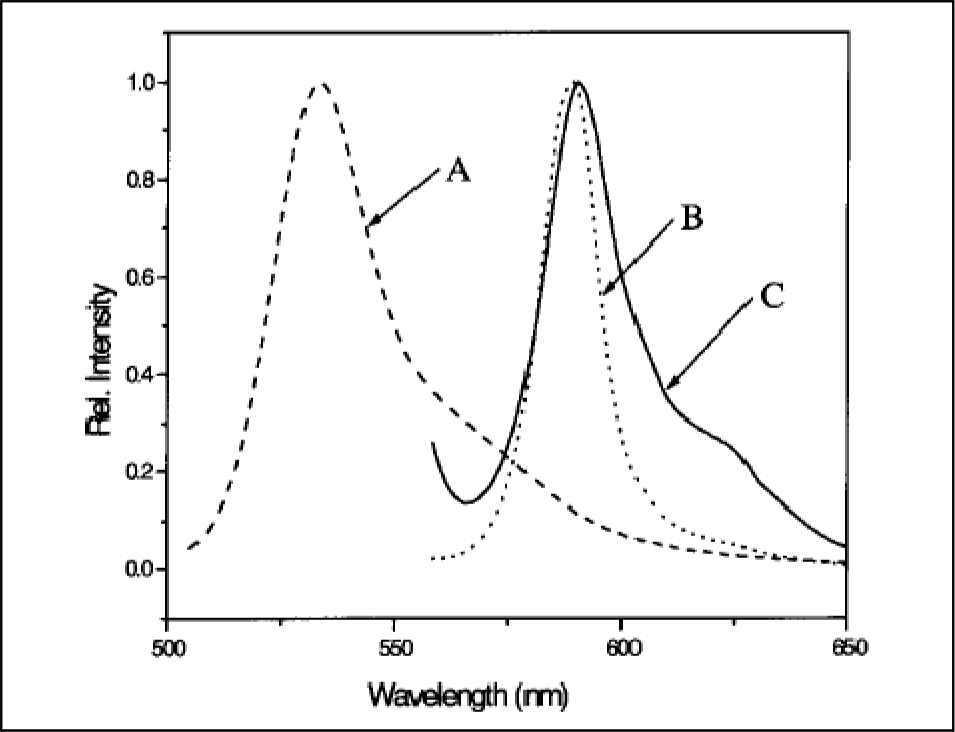
Fluorescence spectra of TDBC: (A) 5×10−5 M monomeric TDBC in methanol, excited at 490 nm (dashed line); (B) aggregated TDBC at 5×10−5 M and pH = 11.0 using water as a solvent, excited at 550 nm (dotted line); and (C) TDBC-A/SBA-15, excited at 550 nm (solid line)
4.2 Use of AERS in other laboratories to explain experimental observations
Here, a discussion is provided of the published research by other groups that report Raman scattering by aggregated molecules in which AERS has been used to rationalize Raman spectra observed under different conditions. Most of the research publications that are discussed have been identified via the use of a citation search, specifically, through the Web of KnowledgeSM search engine. The publications are discussed chronologically from the earliest to the most recent. As mentioned earlier, no attempt is made to be exhaustive regarding the literature; rather, publications that the author believes - through their references and focus areas - provide pertinent information are briefly described.
4.2.1 D. M. Chen, T. J. He, D. F. Cong, Y. H. Zhang and F. C. Liu, “Resonance Raman spectra and excited-state structure of aggregated tetrakis(4-sulfonatophenyl)-porphyrin diacid,” J. Phys. Chem. A, 105 , 3981–3988 (2001). [51]
The authors used time-dependent resonance Raman formulas for the Franck-Condon mechanism to calculate Raman scattering cross-sections. Their calculations indicated that the resonance Raman intensities for the porphyrin studied are very sensitive to the line-width factor (Γ) of the aggregated porphyrin's excitonic transition. In particular, they ascertained that the relative intensities of two low-frequency bands, when normalized with a high frequency mode intensity for the non-resonant and resonant conditions, reveal calculated enhancement ratios that are essentially the same as those determined experimentally (values near 3). They explicitly note that their finding is congruent with the explanation provided by AERS theory that the enhancement of the low-frequency bands derives from a term in the denominator of the Raman intensity expression that becomes small as resonance is approached, leading to an enhancement of the Raman cross-section. They also conclude that, when the resonance Raman (RR) spectra are acquired for the blue-shifted H-band of the aggregate, the lack of a dramatic change in relative intensities of low-frequency versus high-frequency bands derives from the fact that the broad absorption width of the H-band does not allow for a sharp resonance between the excitation frequency and the absorption band frequency.
4.2.2 R. F. Pasternack, S. Ewen, A. Rao, A. S. Meyer, M. A. Freedman, P. J. Collings, S. L. Frey, M. C. Ranen, J. C. de Paula, “Interactions of copper(II) porphyrins with DNA,” Inorganica Chimica Acta, 317 , 59–71 (2001). [52]
The interactions of three cationic water-soluble copper(II) porphyrins with calf thymus DNA (CT) are described. The particular CTs used differed in their peripheral substituents. One of the porphyrins (tetrakis(N-methylpyridinium-4-yl)porphinecopper(II); CuT4) was found to simply intercalate, whereas a second porphyrin (tetrakis(4-N,N′,N″-trimethylanilinium)porphine-copper(II); CuTAP) binds externally, with some limited aggregation under high drug-load conditions. A third porphyrin (trans-bis(N-methylpyridinium-4-yl)diphenyl-porphine-copper(II); t-CuPagg), like the free-base t-H2Pagg from which it is derived, was found to form extended electronically-coupled arrays while bound to the DNA template. A number of experimental approaches, including circular dichromism (CD) have been applied to assess whether potential drug interactions occur via intercalation or groove-binding. The authors observe that aggregation on the DNA template changes the intensity pattern of the porphyrin's resonance Raman spectra, with some low- and high-frequency bands becoming strongly enhanced upon aggregation. They conclude that AERS is thus shown to be a new probe of porphyrin–DNA interactions and that it offers several distinct advantages over other techniques. Chief among these is the structural detail revealed by the analysis of Raman-active vibrational modes, which may help in the description of the inter-chromophore and chromophore–template interactions that determine the aggregate structure.
4.2.3 Grant T. Webster, Don McNaughton and Bayden R. Wood, “Aggregated Enhanced Raman Scattering in Fe(III)-PPIX Solutions: The Effects of Concentration and Chloroquine on Excitonic Interactions,” J. Phys. Chem. B, 113 , 6910–6916 (2009). [53]
Hemes - chemical compounds with iron in the centre of a porphyrin - are commonly recognized as key components of haemoglobin, the red pigment in human blood. The present publication concerned itself with understanding the photophysical behaviour of hemes at high concentrations, with the goal of developing antimalarial drugs that function by impeding the formation of hemozoin, a by-product formed by the catabolism of haemoglobin by the malaria parasite (Plasmodium falciparum), which is concentrated in the digestive vacuole of the malaria parasite. Quinoline antimalarial drugs, such as chloroquine (CQ), are believed to function by targeting Fe(III)PPIX in the form of haematin, its μ-oxo dimer ([Fe(III)PPIX]2O) or the biomineralized hemozoin ([Fe(III)PPIX]2) pellet in the gut of the parasite. The authors utilize resonance Raman and UV-visible absorption measurements to study the effects of various concentrations of CQ in Fe(III)PPIX solutions on structural changes induced upon CQ-Fe(III)PPIX formation. AERS is suggested as being active in Fe(III)PPIX solutions, as indicated by an increase in intensity of A1g modes as a function of concentration and for their excitation wavelength dependence. It is hypothesized that CQ acts as a molecular spacer that binds to the of μ-oxo dimer Fe(III)PPIX units through π–π interactions and other supramolecular interactions, resulting in reduced excitonic interactions, manifested as a lowered intensity of the A1g totally symmetric modes as a function of increasing CQ concentrations. Moreover, UV-visible measurements are interpreted as indicating that CQ binds to the unligated faces of Fe(III)PPIXOH/H2O monomer units, reducing the formation of π–π dimers, as evinced by a red shift in the Soret band as a function of increasing the CQ mole ratio.
4.2.4 D. M. Coles, A. J. H. M. Meijer, W. C. Tsoi, M. D. B. Charlton, J.-S. Kim, D. G. Lidzey, “A Characterization of the Raman Modes in a J-Aggregate-Forming Dye: A Comparison between Theory and Experiment,” J. Phys. Chem. A, 114, 11920–11927 (2010). [54]
The authors investigated the aggregation properties of the TDBC cyanine dye (see Dye-4 in Diagram 2, above) both experimentally and through the use of the Gaussian quantum chemical calculations of three different dimer arrangements. Dimeric systems were used for computational feasibility, with the assumption that the interactions implicit in the dimers would dominate in any aggregate system containing several interacting monomers. Experimentally, an enhancement in the intensity of certain Raman modes upon the aggregation of the dye was observed and the calculated normal mode (with associated atomic displacements for the monomer) suggested that particular out-of-plane motions participated in the enhanced intensity of specific low-frequency Raman bands. The authors explain their findings using AERS.
4.2.5 Ratchadaporn Puntharod, Grant T. Webster, Mehdi Asghari-Khiavi, Keith R. Bambery, Feryal Safinejad, Shadi Rivadehi, Steven J. Langford, Kenneth J. Haller and Bayden R. Wood, “Supramolecular Interactions Playing an Integral Role in the Near-Infrared Raman “Excitonic” Enhancement Observed in β-Hematin (Malaria Pigment) and Other Related Heme Derivatives,” J. Phys. Chem. B, 114 , 12104–12115 (2010). [55]
A number of model compounds were investigated to gain insight into the mechanism that enables the dramatic resonant Raman enhancement of totally symmetric modes observed in hemozoin (malaria pigment) when near-infrared excitation wavelengths (e.g., 782 and 830 nm laser lines) were used for solid state and solution model porphyrin samples. The authors indicate that the fact that the solution-phase spectra evince a large amount of fluorescence when using near-IR excitation provides evidence for AERS in the solid state (where fluorescence is minimized) because the net enhancement of the Raman vibrational modes in this case may derive from the attenuation of fluorescence upon aggregate formation (an indicator of the fact that AERS is occurring). The authors fully embrace the AERS mechanism to explain a number of observations for the system they study.
4.2.6 Christopher C. Rich and Jeanne L. McHale, “Resonance Raman Spectra of Individual Excitonically Coupled Chromophore Aggregates,” J. Phys. Chem. C 117 , 10856–10865 (2013). [56]
The authors speculate that the concept of AERS is valid only if the excitation is resonant with the excitonic transition absorption band. Single aggregate resonance Raman measurements are conducted, which allow non-ensemble information to be acquired. They observe that the significant enhancement of low-frequency modes occurs when the excitation wavelength is in resonance with the J-aggregate absorption band of the aggregate and is presumed to result from the intermolecular nature of the displacements of these modes and their expected coupling to the delocalized electronic transition.
5. Conclusions
As suggested by the discussions above, interest in AERS concepts (by others) as a framework to rationalize Raman scattering by aggregated molecules in their published works have surfaced for a number of very interesting systems in both the physical sciences and the medical research fields. It can be concluded that Raman scattering observations that are consistent with the expectations of the AERS formulation - which exploits molecular exciton theory - are being observed for increasing numbers of systems. The centre of gravity for a sizable fraction of the recent publications that extol AERS concepts appears to be the use of the relative intensities of bands that are enhanced (versus an appropriate internal standard band intensity) when excitation is in resonance with absorption bands. The information extracted through such determinations relates to the relative amounts of aggregated and monomeric structures in the experimental sample. Indeed, many published works point to the use of AERS intensities to determine the minimal amounts of therapeutic agents (in malaria cases) required to disrupt sequestered aggregated materials that, if freed, can enhance the mortality of harmful protozoan parasites that might be injected into red blood cells by mosquitoes. Also, structural alignment information has been acquired by determining which vibrational motions of chromophoric units are in the aggregate formation direction (as indicated by the enhanced intensities of such bands upon resonance excitation). Additionally, in other published works, the determination of which bands experience enhancement -even though the excitation is non-resonant - provides information on the relative alignments of molecular units within the aggregate structure, since even when the exciton is not excited, spatial constriction affects some bands more than others. It is further to be noted that a sizable number of publications continue to look into the fundamentals of the AERS phenomenon.
Of particular note are the potential applications of aggregates occluded within nanoporous materials or nanotubes (i.e., nanoaggregates) which may isolate and protect the active molecular species, thus facilitating the use of such composites: (i) as Raman nanoprobe tags or labels for tracking chemicals, (ii) as bio- and chemo-sensing elements, (iii) as imaging agents, and (iv) as biomarkers to follow disease agents or for drug delivery applications. Increasingly, the Raman scattering of such composites is being used to provide high resolution, finger-print type information.
Footnotes
6. Acknowledgments
Thank is given to the US National Science Foundation for their support of this work under Cooperative Agreement number HRD-08-33180. The author is also deeply indebted to the reviewers for their constructive comments.