
Abstract
Several approaches have been described to identify proteins from MALDI MS/MS mass spectra. The sequence of tryptic peptides is determined by database searching or by de novo sequencing. Different algorithms are available to determine peptide sequence using mass spectra. False discovery of peptides is an associated problem with it. A combination of chemical modifications followed by mass spectral analysis helps in overcoming this problem. Acetylating the tryptic peptides of β-galactosidase in methanol is found to increase the b-ion signal intensity in MALDI TOF mass spectrometry. The method of acetylation is extended to the tryptic peptides of the proteins of an Antarctic bacterium Pseudomonas syringae, whose genome sequence is not known. These proteins are identified by searching the available database of the Pseudomonas spp at NCBI using the MS/MS spectra. The sequences of the peptides are validated using the CID mass spectra of the acetylated tryptic peptides.
Introduction
Sequence determination of tryptic peptides plays a crucial role in the identification of proteins. Collision induced dissociation (CID) mass spectra usually produces intense y ions, with relatively low intense b ion in the spectra. Deducing peptide sequences from fragment ion spectra remains a difficult problem and various approaches are used for the analysis and validation of the data. 1 De novo sequencing is a preferred choice for the determination of peptide sequence when data base is not available or for unknown modified peptides. Several methods have been developed to make this process automated such as Lutefisk 2 Pep Novo 3 novo HMM.4, 5 Incomplete fragmentation patterns and low signal to noise ratio renders it difficult to implement this as a general routine procedure. 6 In addition to the sequence determination of the peptides, chemical modifications followed by detection using mass spectrometry methods have also been used to validate the sequence and identification of proteins.7,8
The sequence of the tryptic peptides has also been identified using database searches, employing several algorithms such as Mascot 9 Sequest 10 and X! Tandem.11, 12 Another way to identify proper sequence of the peptides is in utilizing a combination of database search and de novo sequencing. In this procedure the sequence tag of the peptide is obtained and matched with the results of the database searches. 13 Despite the rapid technology developments and application in many biological studies, the reproducibility of mass spectrometry–-based proteomics has been questioned form time to time.14, 15
Even though several approaches and methods are being developed, the process of determining the sequence of peptides by mass spectrometry is far from complete. It is necessary to develop more methods and approaches for this purpose. Chemical modification of peptides prior to mass spectrometry is another approach for the sequence determination of the tryptic peptides. N-terminal modifications of peptides, such as sodiation, succinylation, acetylation have been carried out in order to increase the relative intensity of a particular type of ions (b ions or y ions) that helps in determining the sequence of peptides.16–20 It has been shown that tryptic peptides upon treatment with N-succinimidyl-2-morpholine acetate exhibited enhanced b ion abundance and facilitated sequence analysis by MS/MS. 21 Picolinamidination of the N-terminal amino group also enhanced b-ion intensity and also facilitated the detection of post translational modifications using MALDI TOF/ TOF.22,23 Most of these studies have been carried out on synthetic peptides and the chemical procedures used for these modifications are hardly suitable to the mixture of tryptic peptides. The chemical reactions described are very laborious to perform while working with hundreds of samples at a time, and also the tryptic peptides are generally present in very small quantities (picomoles to femtomoles) as compared to synthetic peptides. In addition, the un-reacted excess reagents in the reaction mixture lead to decrease in the spectral quality. So, in spite of their occasional success, these methods are far from becoming routine procedures in proteomics. Earlier studies in our laboratory revealed that the relative intensity of b-type ions increased in the case of N- terminal acetylated peptides.24,25
In the present study, chemical modification of the peptide is used to improve the quality of the MS/MS spectra and hence its application as an additional tool to identify the structural features of the tryptic peptides. Using this procedure, it is demonstrated that the acetylation followed by the MS/MS spectra using MALDI TOF/TOF allowed validation of the sequence obtained by data base. The method described in the present study is easy and can be applied to large number of samples as required often in proteomic applications. Tryptic peptides of some proteins obtained from an Antarctic bacterium Pseudomonas syringae, whose genome sequence is not available, have been analyzed after chemically acetylating them.
Materials and Methods
Materials
All the chemicals purchased are of highest purity. The trypsinised β-galactosidase peptides obtained from Applied Biosystems (Foster city, CA) was used as standard. Chemicals such as trypsin, matrix α-cyano-4-hydroxy cinnamic acid, ethanol amine, acetic anhydride, were purchased from sigma chemical co (St. Louis). All solvents like methanol, acetonitrile, dichloromethane used are highest purity and purchased from Spectro Chem (Mumbai, India).
Growth and Culturing Bacteria
The bacterium Pseudomomas syringae Lz4W was isolated from the soil samples of Antarctica in and around Lake Zub. The bacterium can grow between 0 °C to 30 °C. It was grown at 22 °C in Antarctic bacterial medium (ABM) as described earlier. 26 Cells were grown to stationary phase and harvested for further studies. The membrane proteins of this bacterium were prepared as described earlier. 27
Gel Electrophoresis
About 240 µg of the membrane proteins were loaded on immobilized pH gradient (IPG) strip (11cms, pH 5–8, Bio Rad, Hercules, CA) for IEF in the first dimension, followed by SDS-PAGE in 12% acryl amide in the second dimension. The IPG strips were rehydrated in the buffer (8M urea, 2% CHAPS, 50mM DTT, 1% ASB 14 and 2% carrier ampholytes pH 3–10, Bio Rad) for 16 hours in the presence of proteins as described earlier 28 with slight modifications. IEF was carried out in a Protean IEF cell (Bio Rad) cell using the appropriate tray with the initial voltage was set at 5000 Volts at 20 °C for a total of 30000 volt hours. The 2D gels were run stained with coomassie blue. In the 2nd dimension the gel was run using SDS PAGE. 12% acryl amide gel (11 cm x 11 cm, 1 mm thickness) was run at constant voltage 120 V and current set at 20 mA, using a dual vertical slab gel electrophoresis apparatus obtained from Broviga (Chennai, India). 25 mM tris-glycine buffer with 0.1% SDS, pH 8.3 was used as the running buffer.
In Gel Digestion of Proteins
The protocol of Ferro et al 29 was used for digesting the proteins with trypsin with some modifications. Briefly, the gel spots were cut and washed thrice with 25 mM ammonium bicarbonate and acetonitrile (1:1), followed by dehydration with acetonitrile and drying on a speed Vac concentrator. About 10 µl of trypsin (10 µg/ml) was added to these gel pieces and incubated for 16–18hrs. The trypsin digested peptides were extracted in to 50% acetonitrile/water containing 5%TFA. The extract was dried and re dissolved in 50% acetonitrile/ water containing 0.1% trifluoroacetic acid (TFA) for further analysis.
Acetylation
The trypsin digested β-galactosidase peptides were acetylated in different solvents to study the efficiency of acetylation. The acetylation mixture of 3% triethyl amine and 12% acetic anhydride were prepared indifferent solvents namely acetonitrile, chloroform, dichloromethane and methanol. To an aliquot of 2 pico moles of the tryptic digests of β-galactosidse as well as the membrane proteins, 2 µl of the acetyaltion mixture was added, incubated for 10 min and vacuum dried. It was re-dissolved in 8 µl of 50% acetonitrile containing 0.1% TFA and analyzed by MALDI TOF/TOF using HCCA as matrix. A mass difference of 42 Da was observed in each of the peptides after acetylation, indicating that the reaction lead to the formation of mono acetyled derivatives. Additional acetylation sites of peptides are generally indicated by an increase of mass of the peptides by multiples of 42 Da.
Mass Spectrometry
The PMF of different proteins was recorded on a 4800 MALDI TOF/TOF mass analyzer obtained from Applied Biosystems (Foster city, CA). The mass spectra were recorded in reflectron mode using HCCA as matrix. 2 mg of the matrix was dissolved in 1 ml of 50% acetonitrile containing 0.1% TFA. CID mass spectra were recorded for different peptides and their acetyl derivatives using air as collision gas. The mass spectra were recorded in the mass range m/z 800–4000 using fixed laser intensity of 4900 with 2000 laser shots per spectrum. For recording MS/MS spectrum the laser intensity was set at 5900. The first 20 laser shots were always discarded.
Protein Identification
The proteins were identified using PMF and MS/ MS data using NCBI database (updated up to July 2007) containing 99,128 protein sequences. Since the genome sequence of the Antarctic bacterium P.syringae is not known, proteins were identified using the database of the genome sequences of other available Pseudomonas spp as described earlier. 27 Initially PMF and MS/MS spectra of the tryptic peptides have been acquired. After identification of the proteins by NCBI database search, peptides leading to the identification have been listed. From the acetylated samples only those peptides which gave protein identification were chosen for MS/MS studies. The peptide sequences obtained from database searches were validated after acetylating them and observing the b ion intensities.
Results and Discussion
It was proposed to study the effect of acetylation of tryptic peptides on the b-ion intensity in the MS/MS spectra, in order to validate the sequence of the tryptic pepdies. Initial studies were carried out on the tryptic peptides of β-galactosidase. Acetylation of tryptic peptides was carried out in different solvents such as acetonitrile, chloroform, dichloromethane and methanol. Acetylation was found to be most efficient in methanol and mono acetylated peptides were found (Fig. 1). It was also observed that the intensity of the signals of different tryptic peptides was more in methanol as compared to other solvents (Fig. 1A & 1C). The intensity of different peaks were calculated (in %) taking the most abundant peak as 100%. The peptide mass finger print of β-galactosidase and its acetylated peptides are shown in Figure 1. The MS/MS spectra of the tryptic peptides and their acetylated derivatives were recorded. It is observed that the b-ion intensities increased in the acetylated peptide. Figure 1D exhibits the increase in the intensity of b ions of the acetylated peptide of β-gal appearing at m/z 1083.53 corresponding to the sequence GDFQFNISR.
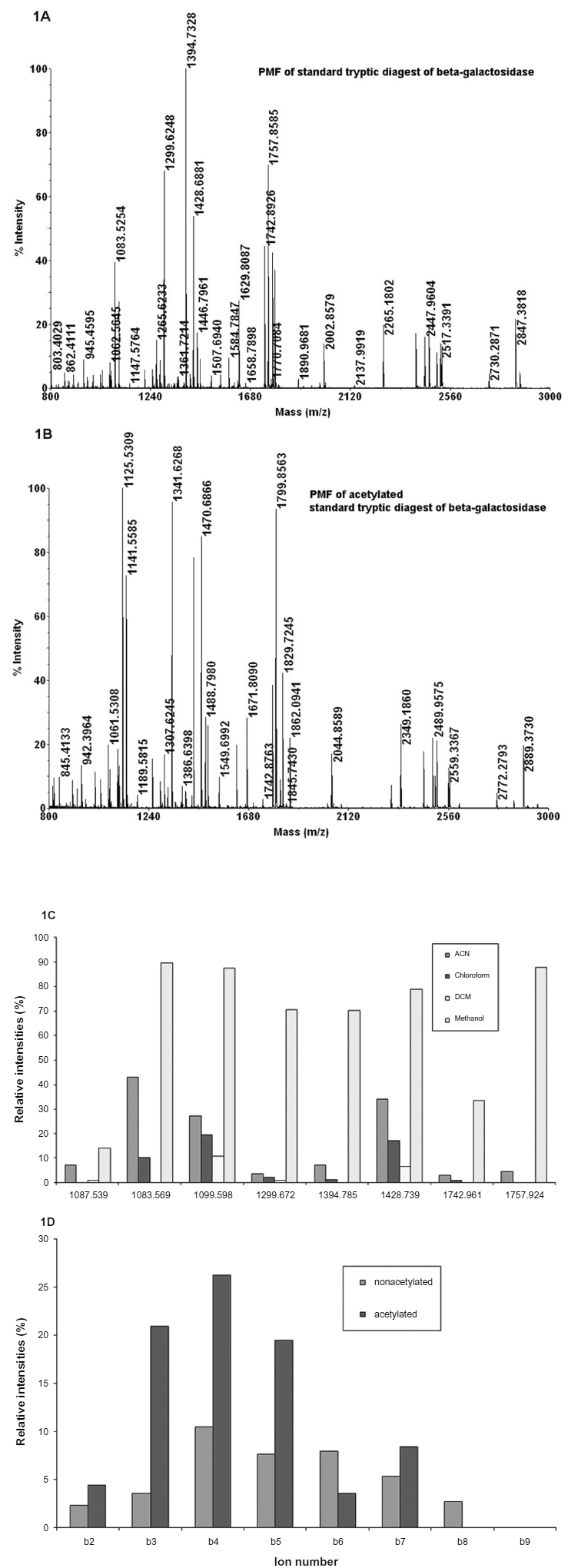
Peptide mass finger print (Panel A) of β-galactosidase from E. coli. Panel B shows the acetylation of tryptic peptides in methanol. Panel C shows the intensity of different tryptic peptides after acetylation in different solvents. The figure shows some representative tryptic peptides and the acetylated derivatives of β-galactosidase appearing at m/z1067.53 (1109.50): WVGYGQDSR, 1083.57(1125.53): GDFQFNISR, 1099.59(1141.55): TDRPSQQLR, 1299.67(1341.62): ELNYGPHQWR, 1394.78(1436.73): LPSEFDLSAFLR, 1428.74(1470.68): DWENPGVTQLNR, 1742.96(1784.89): LSGQTIEVTSEYLFR, 1757.92(1799.85): VNWLGLGPQENYPDR. The mass shift after acetylation was shown in brackets. Panel D shows the b-ion intensities of a peptide GDFQFNISR, before (blue) and after (pink) acetylating it.
Most of the tryptic peptides observed from β-galactosidae show that it contains arginine at the carboxyl terminal, and the N-terminal of the peptide is acetylated. Tryptic peptides containing arginine at the carboxy terminal are more intense in the MALDI spectra than the peptides containing lysine. In the case of peptides containing lysine at the carboxyl terminus, both mono and di- acetylated peptides were observed (Table 1). Peptides containing threonine, serine form additional acetylated derivatives. Thus, the presence of these different residues can be predicted based on the number of acetylated derivatives. Acetylation seems to occur first at the primary amino group present at the N-terminus of the peptide, followed by acetylation at other possible sites. Even if di-acetylated peptides are formed during the reaction, the mono acetylated peptides can be particularly selected for MS/MS spectra. In addition to the immonium ions of the amino acid residues, these acetyl derivatives provide additional information of the type of some individual amino acid residues present in the peptide.
Characteristic features of the acetylated tryptic peptides from P. syringae after MALDI TOF analysis.

M-Oxidized methionine.
Proteins identified using NCBI database search with MS/MS spectra.
The number of b-ions increased in monoacetylated peptides.
The number of acetyl derivatives observed in PMF.
standard sample obtained from Applied Biosystems. ND: Multiple acetylations not detected.
The proteins of the Antarctic bacterium P. syringae are separated on 2D gels to identify the proteins (Fig. 2) using the method described above. Protein spots of different intensities were cut from the gel, and digested with trypsin. Figure 3A & B shows the PMF of DNA binding response regulator Pho B from P.syringae and its acetyl derivatives. The MS/MS spectra of the peptide from this protein appearing at m/z 1562.79, and its acetylated derivative corresponding to the sequence ALGEAYENLVQTVR is shown in Figure 3C & D. The increase in b-ion intensities clearly validate the sequence obtained by data base search (see Supplementary Figs. 1 and 2). It was found that proteins with a wide range of concentrations (based on the intensities of the spots) could be identified and acetylation was carried out on all these tryptic peptides (Table 1). It is observed that the number of acetylated derivatives varied in different peptides (Table 1), which depends on the reaction time, the pH of the medium and the number of acetylating sites present in the amino acid residues of the peptide. Thus it is demonstrated that the sequence of the peptides can be validated using the increased b ion intensities of the tryptic peptides. It is also possible to detect the presence of amino acids in the peptide sequence that produces acetyl derivatives.

Separation of proteins of P. syringae on 2D gel stained with coomasie blue. 240 µg of the protein was loaded on the gel. The proteins of different concentrations digested with trypsin for identification were shown in the figure.
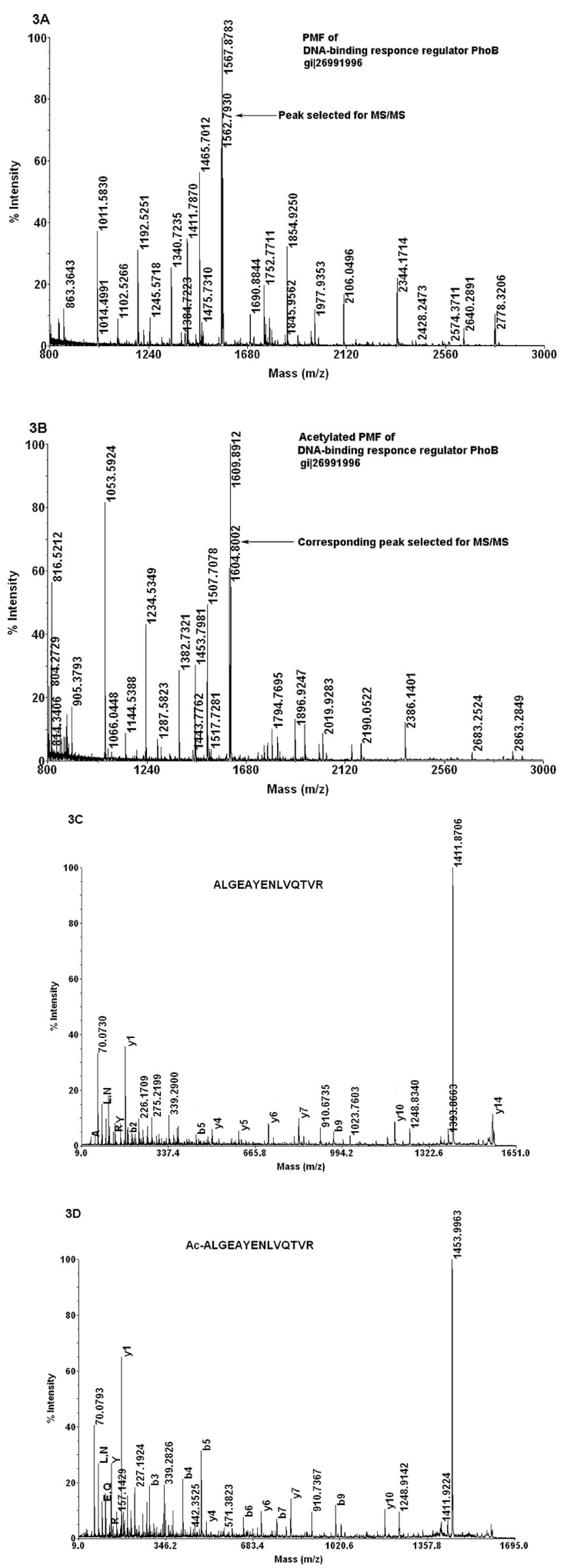
The peptide mass finger print of DNA binding response regulator before (panel A) and after (panel B) acetylation. Panels C and D represents the MS/MS spectra of the peptide ALGEAYENLVQTVR before and after acetylating it. The b ions, y ions and the immonium ions are shown in the figure.
Conclusion
The intensity of b-type ions was found to increase in the MS/MS spectra of mono acetylated trypric peptides. The method for acetylating the peptides has been standardized and extended to large number of tryptic peptides as well as the in-gel digests of protein samples. This method is particularly useful in identifying the proteins from organisms with un-known genome sequence. The increased accuracy in identifying the sequence of peptides from MS/MS spectrum will enable successful design of probes and primers for the genes corresponding to the proteins of interest even when the genome sequence data for the organism in the study is not available. Further studies are in progress to extend this method to sub cellular proteomes as well.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.
Footnotes
Acknowledgement
Financial support from Department of Biotechnology (BT/PR7383/BRB/10/474/2006), New Delhi is acknowledged. HMK and VR are recipients of CSIR research fellowship. We thank Dr. C.S. Sundaram for his help in recording mass spectra, Dr. Sivakama Sundari in the preparation of the manuscript and Ms. Y. Prathyusha for technical help.