
Abstract
Negative feedback regulation of cholesterol metabolism in mammalian cells ensures a proper balance of cholesterol with other membrane lipids, principal among these being the major phospholipid phosphatidylcholine (PC). Processes such as cholesterol biosynthesis and efflux, cholesteryl ester storage in lipid droplets, and uptake of plasma lipoproteins are tuned to the cholesterol/PC ratio. Cholesterol-loaded macrophages in atherosclerotic lesions display increased PC biosynthesis that buffers against elevated cholesterol levels and may also facilitate cholesterol trafficking to enhance cholesterol sensing and efflux. These same mechanisms could play a generic role in homeostatic responses to acute changes in membrane free cholesterol levels. Here, I discuss the established and emerging roles of PC metabolism in promoting intracellular cholesterol trafficking and membrane lipid homeostasis.
Introduction
Cholesterol is an essential lipid component of mammalian cell membranes. Due to its structural properties and assembly characteristics, cholesterol modulates the fluidity of membrane bilayers and partitions into specific lipid domains that are vital to overall cell organization.1,2 The unique rigid ring structure and the small hydroxy headgroup of cholesterol allow it to readily flip-flop between leaflets of a membrane bilayer and to be absorbed and extracted from membranes with relative ease by protein carriers.3,4 Cholesterol is synthesized in the endoplasmic reticulum (ER) and peroxisomes in a multistep process involving more than 20 enzymes and is also released from cholesteryl ester (CE) in cytoplasmic lipid droplets (LDs) or lipoproteins internalized in lysosomes/late endosomes (L/LEs). 3 To maintain membrane cholesterol homeostasis and prevent toxic cholesterol over-accumulation, the cell is equipped with negative feedback mechanisms involving crosstalk between the following two families of sterol-responsive transcription factors: the sterol regulatory element-binding proteins (SREBPs) that control the expression of proteins involved in cholesterol biosynthesis and CE-rich low-density lipoprotein (LDL) uptake 5 and liver-X receptors (LXRs) that increase the expression of cholesterol transporters and promote the efflux of excess free cholesterol to extracellular acceptors. 6 Homeostatic responses to acute changes in cellular cholesterol levels occur mainly in the ER, a cholesterol-poor organelle containing ~1% of total cellular cholesterol. Therefore, interorganelle cholesterol trafficking plays a critical role in cholesterol sensing, yet these mechanisms remain poorly understood.7,8
The heterogeneous distribution of cholesterol in cells is hypothesized to be largely dictated by its affinity for sphingomyelin (SM) and highly saturated phospholipids enriched in lateral membrane nanodomains or rafts in the plasma membrane (PM), trans-Golgi network, and endocytic membranes.9–11 A recent study using high-resolution secondary ion mass spectrometry revealed that micrometer-scale sphingolipid-enriched domains in the PM of cultured fibroblasts were not enriched in cholesterol. 12 This observation should be examined further in other cell types and under different metabolic conditions. It should also be noted that membrane lipid nanodomain size is considered to be in the scale of 10–100 nm,13,14 highlighting the technical challenges in assessing their role in cellular cholesterol distribution. Other phospholipids could also play an important role, such as phosphatidylserine that has recently been shown to affect enrichment of cholesterol in the cytoplasmic leaflet of the PM. 15 It has been postulated that membrane cholesterol in excess of the complexing activity of phospholipids represents the active or free cholesterol capable of dynamic movement, including transport between organelles, conversion to regulatory oxysterols, and efflux to extracellular acceptors. 16 Consistent with this idea, treatment of cells with agents that disrupt membrane domains, such as SMase and oxysterols, results in a redistribution of PM cholesterol to the ER.17,18 The rapid transfer of cholesterol between cell compartments in part involves nonvesicular transport at membrane contact sites (MCSs) that allow cholesterol to freely equilibrate between donor and acceptor membranes. 19 The efficiency of this mode of transport is presumably limited by free cholesterol concentrations in acceptor membranes. Therefore, membrane-driven processes that act as sinks for excess ER cholesterol have the potential to drive cholesterol transport to the ER from other locations. These cholesterol sinks include (1) ER membrane phospholipid biogenesis to absorb free cholesterol, (2) CE storage in LDs, and (3) cholesterol efflux (Fig. 1). In this commentary, I discuss the influence of the major membrane phospholipid phosphatidylcholine (PC) on these processes and thus the potential role of PC metabolic enzymes in promoting intracellular cholesterol transport and membrane cholesterol homeostasis.
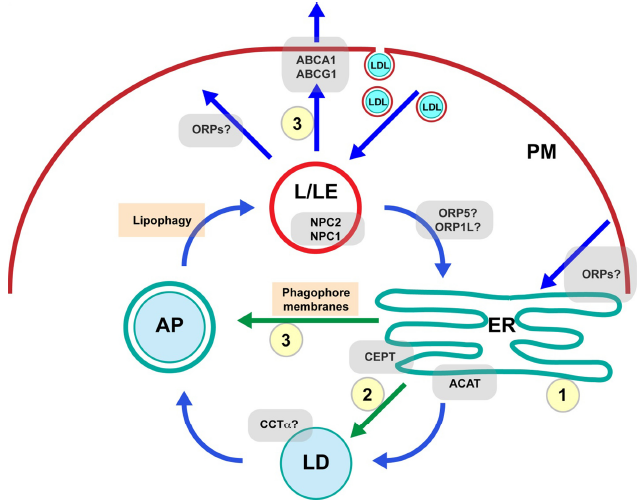
A simplified model of intracellular cholesterol trafficking. PC metabolism potentially regulates several important cholesterol sinks (yellow circles) that maintain low ER cholesterol concentrations. These in turn improve cholesterol sensing and facilitate interorganelle cholesterol trafficking (blue arrows), including pathways to the ER from sites of high cholesterol concentration, such as L/LEs and PM. Trafficking of newly synthesized PC relevant to cholesterol sinks is indicated by green arrows. Cholesterol sinks regulated by PC metabolic enzymes include (1) ER membrane biogenesis and expansion, (2) CE storage in cytoplasmic LDs, and (3) cholesterol efflux involving LD turnover by lipophagy, which may involve PC synthesis in the ER to supply phagophore membranes for autophagosome (AP) biogenesis.
PC Buffers against Excess Cholesterol in Cells
PC is the most abundant phospholipid in eukaryotic cells and has a positive influence on the incorporation of cholesterol in membranes.20,21 PC is also a precursor for the synthesis of SM by the enzyme SM synthase 1 in the Golgi apparatus 22 and, thus, can indirectly affect SM-enriched nanodomains that may affect intracellular cholesterol distribution. 9 PC and cholesterol contents are maintained within narrow ratios in mammalian cells, reflective of the counter-balancing effects of these lipids on membrane fluidity.2,23 As an example, cholesterol-laden macrophage foam cells within atherosclerotic lesions have highly upregulated PC biosynthesis that fuels ER membrane biogenesis to absorb excess free cholesterol.24,25 Failure in this adaptive response results in apoptotic cell death due to toxic concentrations of cholesterol in ER membranes and the resultant ER stress. 26 Under basal conditions, excess free cholesterol is converted to CE by the ER-localized enzyme acyl-CoA:cholesterol acyltransferase 1/2 for storage in cytoplasmsic LDs. 8 LD CE is continuously hydrolyzed and reesterified, which cycles the excess cholesterol through membrane pools for cholesterol sensing and efflux pathways. 27 LDs are dynamic organelles consisting of a neutral lipid core surrounded by a membrane monolayer composed largely of PC. 28 PC biosynthetic enzymes have been shown to regulate the LD size and number in a variety of cell types.29–32 Therefore, PC metabolism regulates not only the capacity of cell membranes to absorb free cholesterol but also the storage of excess cholesterol in LDs.
Enzymes of PC Metabolism
The Kennedy pathway for de novo PC synthesis consists of three enzymatic reactions for the conversion of the soluble metabolite choline to membrane PC. First, choline kinase phosphorylates choline, followed by the formation of the nucleotide intermediate cytidine diphosphate (CDP)-choline catalyzed by CTP:phosphocholine cytidylyltransferase (CCT), and finally the transfer of phosphocholine from CDP-choline to diacylglycerol (DAG) catalyzed by the ER-localized enzyme choline/ethanolamine phosphotransferase 1 (CEPT1).33,34 The rate-limiting reaction under most conditions is the formation of CDP-choline catalyzed by CCT. 35 CCTα, the major ubiquitously expressed isoform, cycles between the nucleus and cytoplasm in response to cellular demands for PC.30,36,37 For instance, CCTα is cytosolic in type II alveolar cells that require large amounts of PC for secretion of the lung surfactant 38 but is predominantly nuclear localized in a variety of other cells and tissues.30,39,40 CCTα is an amphitropic enzyme, existing as both soluble/inactive and membrane-bound/active forms.35,39,41 Membrane translocation occurs via an amphipathic α-helical regulatory domain (domain M) and is triggered by high levels of lipid activators, typically substrates for phospholipid biosynthesis, such as fatty acids and DAG. 42 CCTα also translocates to membrane surfaces that exhibit lipid-packing defects, thus signaling the need for increased PC content.42,43 Binding of CCTα to artificial liposomes causes membrane nanotubule formation, signifying a membrane-deforming ability similar to several other lipid-binding proteins containing amphipathic helical domains.44,45 Within cells, nuclear CCTα membrane translocation induces the formation of tubular invaginations of the double membrane nuclear envelope, collectively termed the nucleoplasmic reticulum, implicated in intranuclear calcium signaling and nucleo/cytoplasmic transport.44,46 The membrane-deforming ability of CCTα could also impact extranuclear sites of membrane translocation, but this possibility remains unexplored.
The Kennedy pathway is responsible for de novo PC production in all cells and tissues, with the methylation of phosphatidylethanolamine (PE) to PC catalyzed by PE methyltransferase also playing a major role in liver and adipocytes. 47 Further metabolism of de novo synthesized PC through the Lands cycle involves fatty acid removal from the sn-2 position by phospholipase A2 activity followed by reacylation by lysophosphatidylcholine acyltransferase (LPCAT) that modifies the fatty acid composition of PC to the mature form found in membranes.48,49 Stimulated PC turnover by various phospholipases produces bioactive lipids, such as phosphatidic acid, lysophosphatidylcholine, DAG, and unsaturated fatty acids. 33 PC synthesis and turnover are closely regulated with the cell cycle, thus ensuring the coordination of cell growth and membrane biogenesis. 33 In other conditions of stimulated membrane growth, such as ER stress-induced ER expansion, increased flux through the Kennedy pathway can produce an overshoot response.50,51 However, studies of CCTα overexpression or increased activation indicate that excess PC in cells is efficiently degraded to maintain steady-state membrane levels.52,53
The Role of PC in Cholesterol Storage in LDs
Due to the ubiquitous role of PC in membrane structure and as a source of bioactive lipids, defining the role(s) of PC metabolism in cholesterol trafficking is complex. An emerging area of interest involves the role of PC in regulating the function of LDs, metabolic organelles that control membrane lipid storage and trafficking.
54
The esterification of excess cholesterol and fatty acids for storage in LDs occurs in all cell types and plays a critical role in membrane homeostasis.
55
LDs consist of a neutral lipid core of triacylglycerol (TAG) and CE surrounded by a membrane monolayer composed mainly of PC with numerous associated proteins.54,56 LDs are dynamic organelles that rapidly change their size, number, and position in the cytoplasm depending on the nutritional status of the cell.
28
A genome-wide RNAi screen conducted in oleate-loaded
PC and Cholesterol Coregulation by the SREBP Pathway
The central regulators of membrane lipid homeostasis are the SREBPs, a family of transcription factors that regulate the expression of genes involved in cholesterol and fatty acid synthesis and their uptake in plasma lipoproteins. 5 The SREBP isoforms have the following separable but overlapping gene targets: SREBP-1a controls genes in both cholesterol and fatty acid biosynthetic pathways, while SREBP-1c, the major SREBP-1 isoform expressed in tissues, and SREBP-2 are relatively specific for fatty acid and cholesterol synthesis, respectively. 61 SREBPs are initially synthesized as inactive membrane-bound precursors localized in the ER. Vesicle-mediated transport to the Golgi apparatus results in sequential cleavage by two Golgi-resident proteases, releasing cytosolic mature forms that traffic to the nucleus and activate target genes. 5 Processing of SREBPs is regulated by the cholesterol/phospholipid ratio in ER membranes, where a switch-like change in the chemical activity of cholesterol occurs above a threshold of ~5 mol%. 62 The cholesterol sensor is SREBP-cleavage-activating protein (SCAP) that binds to and escorts SREBPs from the ER to the Golgi. 63 Direct binding of cholesterol to SCAP induces a conformation change and binding to insulin-induced gene protein (Insig). 64 This results in retention of the SCAP/SREBP complex in the ER, thus preventing the proteolytic activation of SREBPs at the Golgi. 65
Differential regulation of SREBP-1 and SREBP-2 isoforms occurs in cultured cells and in liver where SREBP-1c is highly upregulated as compared to SREBP-2 at both the mRNA and processing levels in response to insulin signaling.66,67 Unsaturated fatty acids were shown to be required to fully suppress the processing of SREBP-1 isoforms but not SREBP-2 in the presence of sterols in HEK 293 cells.
68
Recently, it was demonstrated that PC synthesis via the Kennedy pathway negatively affects the proteolytic activation of SREBP-1 isoforms in
Regulation of PC Synthesis by Oxysterols and the LXR Pathway
A portion of excess cholesterol in cells is channeled to mitochondria for the synthesis of various oxysterols with altered physiochemical properties compared to cholesterol, such as enhanced solubility and spontaneous diffusion across cellular membranes.74,75 The majority of cholesterol turnover in the body takes place in the liver where 7α-hydroxylated sterols are substrates for the synthesis of bile acids. 76 In the brain, turnover of excess cholesterol involves its conversion to 24-hydroxycholesterol, followed by secretion and clearance from the circulation by the liver for incorporation into the bile acid biosynthetic pathway.77,78 In all cells and tissues, oxysterols serve as potent signaling molecules that improve cell and whole body membrane homeostasis through several mechanisms. 74 For instance, oxysterols induce degradation of 3-hydroxy 3-methylglutaryl (HMG)-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis, and suppress cholesterol biosynthetic gene expression by inhibiting SREBP-2 proteolytic activation.79,80 Although cholesterol itself is a poor membrane activator of CCTα, the oxysterol 25-hydroxycholesterol directly stimulates CCTα activity in vitro and induces CCTα membrane translocation and activation in cultured cells. 81
A primary signaling function of oxysterols is as ligand activators of LXRs (LXR-α and LXR-β), transcription factors that increase the expression of cholesterol transporters, such as the ATP-binding cassette transporter ABCA1, along with other proteins involved in cholesterol efflux. 82 Through a process known as reverse cholesterol transport (RCT), excess cholesterol from peripheral tissues is effluxed to high-density lipoprotein acceptors and transported to the liver for eventual excretion in the bile. The protective effects of RCT are especially vital for the removal of cholesterol from the intimal space of the vasculature, where macrophages are prone to becoming atherogenic foam cells through the scavenging of oxidized LDL particles. 83 Accordingly, LXRs have emerged as important therapeutic targets to inhibit atherosclerosis by promoting RCT. 84 However, a caveat is that SREBP-1c is also an LXR target gene. As a result, treatment with synthetic LXR agonists leads to a detrimental increase in liver lipogenesis accompanied by hypertriglyceridemia and hepatic steatosis in animal models tested. 85 The role of SREBP-1c induction in the LXR gene regulatory program remains unclear. As noted earlier, SREBP-1c activity produces membrane activators of CCTα along with substrates for de novo PC synthesis. 70 Thus, one possibility is that increased lipogenesis under conditions of excess cholesterol improves the ratio of free cholesterol with membrane phospholipids. 86 The increased fatty acid and TAG production could also detoxify the effect of elevated cholesterol by promoting its storage as CE in cytoplasmic LDs. 87
Potential Role of Membrane PC Synthesis in Lipophagy
Autophagy is a constitutive degradative pathway conserved from yeast to humans that plays an essential role in the maintenance of cellular energy and metabolic homeostasis. Autophagic (self-eating) degradation involves engulfment of targeted cytosolic components by phagocytic membranes to form an autophagosome (AP) that fuses with L/LEs where the contents are degraded, releasing nutrients and other building blocks for use in cellular metabolism. 88 A specialized form of autophagy known as lipophagy degrades cytoplasmic LDs, thus liberating stored fatty acids for energy production during starvation. 89 Recently, lipophagy was shown to play a critical role in cholesterol efflux from macrophage foam cells, whereby lysosomal acid lipase hydrolyzes LD CE releasing free cholesterol for ABCA1-mediated efflux. 90 Membranes required for AP formation can arise from various sources depending on the cellular context. In starvation-induced macroautophagy, this may involve physical remodeling of existing membranes originating at the ER, 91 ER–mitochondria contact sites,92,93 Golgi apparatus, 94 or recycling endosomes.95,96 Under nutrient replete conditions, AP biogenesis has been shown to involve hydrolysis of TAG stores in LDs, releasing fatty acids to supply de novo PC synthesis in the ER. 97 Formation of APs to engulf large CE-rich LDs may also require lipogenesis to provide the needed substrate for new membrane lipid synthesis. Thus, SREBP-1c induction leading to CCTα activation and increased de novo PC synthesis could support lipophagy, identified as an essential early step in LXR-stimulated cholesterol efflux from macrophage foam cells. 90 While LD degradation by lipophagy has been demonstrated in extreme conditions, such as starvation 89 and cholesterol-laden macrophage foam cells, 90 a generic role in membrane homeostasis remains undefined. Importantly, a recent report showed that SREBP-2 regulates the expression of several protein factors involved in autophagy. 98 This points to a potential role of lipophagy in mobilizing LD CE stores to augment membrane free cholesterol pools under cholesterol-depleted conditions, allowing a more rapid response as compared to CE hydrolysis by a cytoplasmic neutral CE hydrolase. If so, the potential roles of PC metabolism in AP formation and lipophagy may not only apply to extreme situations of cholesterol loading in cells but also to cholesterol sensing and membrane cholesterol homeostasis under basal conditions.
Transport of Cholesterol from L/LEs
An important cholesterol trafficking pathway originates in L/LEs where hydrolytic breakdown of the CE component of internalized LDL releases free cholesterol for use in cell metabolism.
3
PM cholesterol internalized in endocytic vesicles is also processed in the L/LE compartment for recycling to intracellular membranes.
99
An initial step is the binding of free cholesterol by the soluble luminal protein Niemann–Pick C (NPC) 2, followed by handoff of the cholesterol molecule to the polytopic membrane protein NPC1 for transfer to unknown cytosolic binding proteins or insertion into the L/LE-limiting membrane.
100
NPC disease is a progressive fatal disorder associated with neurodegeneration and liver dysfunction caused by loss-of-function mutation in either
The cholesterol trafficking pathways from L/LEs to other organelles remain largely unknown but involve both vesicular and nonvesicular mechanisms.3,107,108 Potentially important players are members of the oxysterol-binding-protein (OSBP)-related protein (ORP) family, known to participate in nonvesicular transfer of sterols and other lipids at interorganelle MCSs.109,110 OSBP along with several other cytoplasmic ORPs binds to oxysterols, which induces membrane translocation in conjunction with specific organelle-targeting signals. 111 Several ORPs have recently been implicated in the nonvesicular export of cholesterol from L/LEs. In one study, ORP1L was shown to interact with the ER resident protein, vesicle-associated membrane protein-associated protein, to regulate L/LE positioning in response to cholesterol depletion and L/LE-to-ER MCSs in human MelJuSo melanoma cells. 112 Knockdown of ORP5, a membrane-anchored ORP localized in the ER, caused L/LE accumulation of LDL-derived free cholesterol in HeLa cells, mimicking the NPC phenotype. 113 Detailed confocal microscopy analysis revealed that accumulation of free cholesterol occurred in the L/LE-limiting membrane in addition to the luminal compartment. 113 Therefore, ORP5 may function downstream of NPC1 to mediate the exit of LDL-derived cholesterol from L/LEs to the ER. A recent study showed that L/LE-to-peroxisome transport of cholesterol occurs via a MCS involving the L/LE membrane protein synaptotagmin VII binding to peroxisomal PI(4,5)P2. 108 Knockdown of critical peroxisomal genes in HEK 293 cells blocked L/LE cholesterol trafficking and caused L/LE accumulation of LDL-derived cholesterol. In addition, L/LE cholesterol accumulation occurred in cell and animal models of various peroxisomal disorders. 108 Importantly, these results point to a previously unappreciated role for peroxisomes as an important conduit for L/LE cholesterol export and, further, that this transport is mediated through MCSs. 108
Potential Roles of Cholesterol Sinks in NPC Disease Reversal
Various manipulations in lipid metabolism or membrane trafficking have been shown to reverse L/LE cholesterol accumulation in NPC1-deficient cells.114–119 Several of these mechanisms appear to act at extra-lysosomal sites and, thus, may relieve bottlenecks in the intracellular movement of cholesterol. Conversely, the accumulation of lipoprotein-derived cholesterol in L/LEs has been shown to occur in cells overloaded with free cholesterol in the absence of NPC protein defects.120,121 This suggests that the export of free cholesterol from L/LEs becomes rate limiting upon saturation of cholesterol levels in downstream membranes. If, as recent studies suggest, a substantial amount of intracellular cholesterol trafficking involves transport down a chemical concentration gradient at interorganelle MCSs,19,110,122 then lower cholesterol concentrations in acceptor membranes would facilitate cholesterol movement. In this model, mechanisms that lower ER cholesterol concentrations would drive cholesterol trafficking to the ER from distal sites, leading to improved cholesterol sensing and homeostasis. As detailed herein, PC metabolism is intimately linked with several cholesterol sinks that may facilitate cholesterol trafficking to the ER by this mechanism, specifically (1) expansion of ER membranes to absorb free cholesterol, (2) LD CE storage, and (3) cholesterol efflux (Fig. 1). Increased flux in these pathways could conceivably alleviate the cholesterol trafficking block in NPC1-deficient cells by improving the kinetics of NPC2-mediated cholesterol transfer. Indeed, one study showed that ABCA1 overexpression reversed the NPC disease phenotype in human fibroblasts deficient in NPC1 but not in NPC2. 117 This effect was highest upon addition of the cholesterol efflux acceptor apoA-I to the culture medium, suggesting that increased ABCA1-mediated cholesterol efflux provided the needed cholesterol sink to bypass the requirement for NPC1. However, it is also possible that the overexpressed ABCA1 acted directly in cholesterol transfer at the L/LE membrane in cooperation with NPC2. 117 Clearly, further studies are needed to investigate whether reversal of the cholesterol trafficking defect in NPC1-deficient cells and tissues requires treatments that compensate directly within the L/LE compartment or whether downstream cholesterol sinks can shift the rate-limiting step and improve the kinetics of NPC2-dependent cholesterol transfer. In this regard, the role of PC metabolism in L/LE cholesterol export could be tested in cell models in which PC synthesis is upregulated and/or PC turnover is inhibited, leading to an increase in the cellular PC/cholesterol ratio of downstream membranes. Conversely, knockdown of key enzymes in the Kennedy pathway, such as CCTα and CEPT1, could lead to decreased trafficking of LDL-derived cholesterol from L/LEs and mimic the NPC phenotype.
Conclusion
The maintenance of cholesterol/PC ratios in cell membranes is vital to membrane integrity and fluidity and promotes stability and function of transmembrane proteins. Critical increases in ER cholesterol concentrations result in depletion of ER calcium stores and activation of ER stress pathways that can lead to apoptotic cell death. 26 Sequestration of excess free cholesterol in L/LEs could protect the cell against cholesterol toxicity and ER stress in the short term, but the long-term consequence is pathological L/LE dysfunction, as seen in various neurodegenerative lipid storage disorders. 123 L/LE cholesterol accumulation in tissue macrophages in the vessel wall and liver has also been linked to detrimental inflammatory responses in cardiovascular disease and nonalcoholic steatohepatitis, respectively.124,125 PC plays a critical role in cellular cholesterol sinks that buffer against cholesterol-induced ER stress and assist in the maintenance of cellular cholesterol gradients that drive interorganelle cholesterol transport. Therefore, the enzymes of PC metabolism potentially represent control points affecting cholesterol trafficking and storage and, ultimately, cholesterol homeostasis.
Author Contributions
Conceived the concepts: TAL. Wrote the first draft of the manuscript: TAL. Developed the structure and arguments for the paper: TAL. Made critical revisions: TAL. The author reviewed and approved of the final manuscript.