
Abstract
In the mammalian central nervous system, reactive oxygen species (ROS) generation is counterbalanced by antioxidant defenses. When large amounts of ROS accumulate, antioxidant mechanisms become overwhelmed and oxidative cellular stress may occur. Therefore, ROS are typically characterized as toxic molecules, oxidizing membrane lipids, changing the conformation of proteins, damaging nucleic acids, and causing deficits in synaptic plasticity. High ROS concentrations are associated with a decline in cognitive functions, as observed in some neurodegenerative disorders and age-dependent decay of neuroplasticity. Nevertheless, controlled ROS production provides the optimal redox state for the activation of transductional pathways involved in synaptic changes. Since ROS may regulate neuronal activity and elicit negative effects at the same time, the distinction between beneficial and deleterious consequences is unclear. In this regard, this review assesses current research and describes the main sources of ROS in neurons, specifying their involvement in synaptic plasticity and distinguishing between physiological and pathological processes implicated.
Introduction
Reactive oxygen species (ROS) are chemically reactive molecules derived from the reduction of molecular oxygen. In living organisms, ROS are generated as a subproduct of cellular metabolism or through the activity of specific enzymatic complexes. The first step in ROS production is the reduction of molecular oxygen (O2) to anion superoxide (O2−), which is the precursor of other reactive species. Hydrogen peroxide (H2O2) is produced through dismutation (a chemical process that generates oxidized molecules) of superoxide. H2O2 can be converted into hydroxyl radical (OH) or water. 1 OH is extremely reactive and immediately removes electrons from other molecules, turning them into new radicals and propagating the formation of ROS in a chain reaction. H2O2 has lower reactivity, and this condition allows this molecule to diffuse into the cytoplasm and eventually reach the nucleus to interact with DNA.
Over the past decade, the study of the biological activity of ROS in the nervous system has gained particular interest. Since ROS are highly unstable and reactive molecules, they are potentially capable of interfering with many cellular processes. ROS can react with proteins and nucleic acids and disrupt their cellular functions. Typically, oxidative stress occurs when the damaging effects of ROS exceed the ability of biological systems to neutralize the oxidizing agents and repair cellular damage. 2
Nevertheless, the idea that ROS are only harmful molecules that threaten cellular health has been reviewed in recent years. Neurons of the central nervous system (CNS) process neural signals through modification mechanisms of synaptic efficacy.3, 4 These mechanisms require complex intracellular signalization pathways that involve the production of ROS.5, 6 Thus, the relationship between ROS production and synaptic plasticity is extremely ambiguous: ROS appear to be essential for the intracellular signaling involved in plasticity in the CNS,7–12 but at the same time, excessive ROS accumulation in the brain can result in cellular oxidative damage. The boundary between the positive and negative effects of ROS is still unclear, as oxygen metabolism in cells often induces both physiological and pathological consequences, by using intertwined transduction cellular pathways. 13
This review provides an account of the ROS studies undertaken so far in order to provide a clearer understanding. To this end, we discuss the relationship between ROS and plasticity, exposing the main cellular mechanisms responsible for pathological and physiological processes.
Synaptic Plasticity
One of the most remarkable and important features of the CNS is its ability to process and store information through synaptic changes. The capability of synapses to alter their own strength as a response to previous stimulation is called synaptic plasticity. 14 Synaptic modifications result from changes in the quantity of neurotransmitters released and/or from changes in how effectively the cells respond to neurotransmitters. Such modifications comprise long-term potentiation (LTP), which is a long-lasting increase in synaptic efficiency, and long-term depression (LTD), which is a long-lasting decrease in the strength of synaptic transmission.15–18 The level of intracellular calcium (Ca2+) is the key factor triggering LTP or LTD in excitatory glutamatergic neurons.19, 20 During the induction of synaptic plasticity, intracellular calcium levels may increase due to stimulation of N-methyl-D-aspartate (NMDA) receptors, opening of L-type voltage-dependent calcium channels (L-VDCCs), and activation of metabotropic glutamatergic receptors.
In the hippocampus and cerebral cortex, high-frequency stimulation (HFS) results in massive calcium influx, inducing LTP, while low-frequency stimulation (LFS) leads to lower calcium currents and triggers LTD.16, 21–25 In these structures, NMDA glutamate receptors are the principal factors enabling the rise of intracellular Ca2+ inside the postsynaptic neuron. NMDA receptor activation requires both presynaptic glutamate release and depolarization of the postsynaptic plasma membrane. When HFS of presynaptic fibers occurs, high amounts of glutamate are released and activate alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) postsynaptic glutamatergic receptors, inducing strong membrane depolarization. Such levels of postsynaptic depolarization displace the magnesium ions that normally block the NMDA receptor channel. Thus, HFS presynaptic fiber leads to strong NMDA receptor activation, providing high calcium influx in the postsynaptic neuron. On the other hand, LFS leads to lower levels of postsynaptic depolarization, and therefore, the calcium influx passing through the NMDA receptor is lower compared to HFS. The calcium signals caused by HFS lead to LTP expression, while the amount of calcium provided by LFS induces LTD.18, 26–28 Since NMDA receptors are sensitive to both presynaptic glutamate release and postsynaptic depolarization, they act as coincidence detectors of pre-and postsynaptic activities. 29 The probability of NMDA receptors opening is directly related to the frequency of stimulations being delivered to synapses. In this way, synapses can selectively strengthen or weaken in a use-dependent manner.
Once calcium has entered the postsynaptic cell, the expression of synaptic plasticity requires the activation of multiple protein kinase signaling cascades, such as the calcium calmodulin kinase II (CaMKII), extracellular signal-regulated kinase (ERK), cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA), and protein kinase C (PKC).3, 30 These pathways modify the number and efficacy of AMPA glutamate receptors in the postsynaptic membrane, allowing changes in the glutamate-induced depolarization. After LTP is induced, the number and efficiency of AMPA receptors increase, while the opposite occurs during LTD.3, 30
Often, the long-term maintenance of synaptic changes requires the synthesis of new proteins.31, 32 Intracellular pathways activated by Ca2+ induce nuclear translocation of specific kinases (PKA, CaMKII, and ERK), which in turn phosphorylate other targets, including transcription factors such as the cAMP response element-binding protein (CREB). The combined activation of the abovementioned transduction pathways results in multiple transcription factors initiating a wave of transcriptions. This process results in newly formed proteins, such as the AMPA receptor sub-units GluR1 and GluR2, brain-derived neurotrophic factor (BDNF), tissue plasminogen activator, postsynaptic density protein 95 (PSD-95), and fragile X mental retardation protein. These new proteins allow the possibility of long-term synaptic modifications by altering the electrical properties of the membrane, changing receptor expression, modifying synaptic morphology and size, and changing the number of synaptic connections. 33
Thus, in forebrain circuitries, high NMDA receptor-dependent increases in intracellular calcium are commonly associated with LTP induction, while moderate calcium influx is thought to induce LTD. This strict rule does not apply to all SNC structures. For example, in cerebellar synaptic plasticity, high calcium concentrations are responsible for triggering LTD, while LTP requires lower calcium levels. Coupled activation of parallel and climbing fibers provides the calcium concentration required for LTD. Activation of climbing fibers induces Ca2+ influx through voltage-gated Ca2+ channels, while parallel fiber inputs induce Ca2+ release from the intracellular stores by activating the type 1 metabotropic glutamatergic receptor (mGluR1).34, 35 The two Ca2+ signals act synergistically to activate PKC, which is fundamental for the expression of cerebellar LTD. 36 When lower calcium signals stimulate the Purkinje postsynaptic cell, LTP is induced through the activation of the phospholipase A2-arachidonic acid (PLA2-AA) pathway and the production of nitric oxide (NO). 37
Intracellular calcium concentration alone is not the only factor responsible for the occurrence of synaptic plasticity. Spatial distribution and timing of Ca2+ signals also play a role in the expression of plastic changes.38–40 Ca2+ spikes and Ca2+ release from intracellular stores can occur with different timing at different points of dendritic arbors and such differences may result in the activation of different molecular pathways. 41
Synaptic plasticity processes in the hippocampus, cerebellum, and cerebral cortex are probably the most studied, given their obvious relationship with memory and learning. However, the possibility of changes in synaptic transmission is a fundamental property present in most, if not all, structures of the CNS. For example, the phrenic nerve of the spinal cord undergoes synaptic changes to sustain rapid and strong increases in respiratory motor output as a defense against hypoxia. 42 Repeated stimulation of the carotid sinus nerve augments phrenic inspiratory activity through a process of synaptic potentiation called phrenic long-term facilitation (pLTF), which persists despite the regulation of arterial blood gases at their baseline values. 43 Drugs that interfere with serotonergic neurotransmission attenuate this effect, suggesting that LTF is a central neural mechanism of synaptic facilitation and not the simple result of changes in peripheral chemoreceptor sensitivity. 44 This form of plasticity requires the synthesis of BDNF and the activation of the tropomyosin receptor kinase B. 45 Similar to LTP, pLTF involves the stimulation of NMDA receptors as well as the phosphorylation of ERK and PKC.45–47
It has been proposed that age-related cognitive impairments are caused by a decline in synaptic plasticity in neurons of the brain. 48 Neurodegenerative processes are typically characterized by synaptic damage, and synaptic loss is one of the strongest correlates to the cognitive impairment of patients with neurological disorders.49, 50 Several lines of investigation support the notion that such pathologies result in the alteration of intracellular signaling pathways related to synaptic plasticity.51–53 In this sense, investigations into the relationship between ROS and neuroplasticity could provide important perspectives in the understanding of CNS pathologies, considering the ambiguous role of ROS in cellular functions. Increased evidence has shown that ROS act as small signaling molecules triggering the aforementioned intracellular cascades.54–57 Many of the kinase proteins inducing synaptic structural and functional changes require a certain redox environment, as their activity can be modulated by virtue of ROS levels. Modifications in ROS concentrations might serve to regulate the expression of LTP and LTD acting at different steps of the transduction process. In the following sections, we will first describe the cellular structures that are active in the generation of ROS. Then, we will discuss how ROS are involved in synaptic plasticity, both under physiological and pathological conditions.
Sources of ROS in the Brain
Mitochondria are the main cellular organelle involved in the production of energy using oxygen, and for this reason, they produce the largest amount of ROS. Amounts of ROS that are large enough to participate in cellular processes can be generated by other sources, such as the enzyme neural nitric oxide synthase (nNOS) and the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase. In the following sections, we briefly describe the principal sources contributing to the generation of ROS in neurons.
Mitochondria
Neurons use mitochondrial oxidative phosphorylation to generate adenosine triphosphate (ATP), in order to provide the energy required for cellular metabolism. The major byproduct of this process is the anion superoxide, which is rapidly dismutated to H2O2 by the mitochondrial enzyme superoxide dismutase 2 (SOD2). 58 Since neuronal tissues have extremely high metabolic rates, neurons produce elevated amounts of ROS compared to other organs. 59 The production of ROS in the mitochondrial respiratory chain derives from a leak of superoxide in complexes I and III. 60 Complex I operates by pumping four protons from the internal matrix to the intermembrane space, creating the proton gradient necessary for ATP formation. 61 This complex oxidizes the nicotinamide adenine dinucleotide hydrogen (NADH) molecule by using the coenzyme Q10 as an electron acceptor. 60 In this process, electrons are transferred from NADH to molecular oxygen, resulting in the formation of the free radical superoxide. 62 Complex III participates in the generation of the proton gradient, by releasing protons across the mitochondrial membrane. This mechanism requires a two-step reduction of ubiquinonic structures: in the first step, two electrons are removed from the Q0 ubiquinone and transferred to two molecules of cytochrome c; in the second step, two electrons are used to reduce the quinone to quinol. 60 Like complex I, complex III can leak some electrons and contribute to mitochondrial superoxide formation. 63 Mitochondrial ROS generation seems to reflect the level of neuronal activity, as superoxide production is enhanced during intense synaptic transmission.
Mitochondria-derived ROS concentrations are regulated by intracellular calcium levels. ROS increase when mitochondria are treated with high concentrations of Ca2+ and Na+, for example, after sustained NMDA receptor activation.64, 65 Inhibitors of complexes I and III reduce the NMDA receptor-dependent superoxide generation in cortical neurons, suggesting a causal relationship between the NMDA receptor, the electron transport chain activity, and the production of ROS. 66 Ca2+ influx from NMDA receptors triggers mitochondrial activation of caspase-3, which in turn stimulates the synthesis of the myocyte enhancer factor 2 (MEF2). MEF2 regulates the transcription of the mitochondrial gene NADH dehydrogenase 6 (ND6), which encodes an essential component of complex I. When MEF2 expression is blocked, the activity of complex I is decreased, and, consequently, ROS production from the electron transport chain is increased.67, 68 The MEF2-dependent expression of ND6 also reduces cellular levels of the antioxidant enzymes superoxidase and hydrogen peroxidase.
Neural nitric oxide synthase
Nitric oxide synthase (NOS) is an enzyme, which catalyzes the production of NO by oxidizing L-arginine to L-citrulline. NOS activity requires NADPH, tetrahydrobiopterin, and molecular oxygen as cofactors. 69 NO is a free radical with an unpaired electron and is active as a cellular signaling molecule modulating several physiological processes, including immune defense, vascular tone, insulin secretion, peristalsis, airway tone, neural development, and angiogenesis. 70 Although NO is considered a nitrogen reactive species, it is capable of interacting with ROS and interfering with redox homeostasis. NOS can directly interfere with ROS production: once produced, NO can react with superoxide and generate peroxynitrite, a highly reactive compound. 71 Furthermore, if L-arginine is present at low levels, superoxide and H2O2 can be generated instead of NO. 72
The NOS isoform localized in neurons of the nervous system is named nNOS.73, 74 Intracellular-free calcium concentration regulates nNOS through the Ca2+-binding protein calmodulin. During calcium influx inside the cell, NOS become fully active upon interaction with calmodulin. 75 The binding of calmodulin with intracellular Ca2+ becomes the main regulator of nNOS activity. 74 The predominant splice variant of nNOS in the brain contains an N-terminal-binding domain that anchors this complex to the postsynaptic membrane, which is close to the NMDA receptor. 76 This interaction allows the Ca2+ influx through NMDA to be coupled to NO synthesis and activity. 77
Functionally, nNOS represents an important factor regulating synaptic transmission. NO is a critical signaling molecule important for some mechanisms of synaptic plasticity, functioning as a retrograde neurotransmitter during LTP.78, 79 Inhibitors of NOS, competing for the L-arginine substrate, can prevent the induction of LTP. 78
In addition to promoting plasticity, nNOS may act as a factor limiting high levels of NMDA receptor activity. The NMDA receptor function can be blocked by treating neurons with NO-producing agents.80, 81 When nNOS is stimulated with L-arginine, a strong and long-lasting inhibition of the NMDA receptor function is observed.
82
The nNOS-mediated inhibition of the NMDA receptor is suppressed when hemoglobin is adopted as a NO scavenger.
83
NO produced by nNOS inhibits NMDA receptors through mechanisms of
Monoamine oxidase
Monoamine oxidase (MAO) is an enzyme that catalyzes the oxidation of monoamines. Two subtypes of MAO have been described, MAO-A and MAO-B, and both isoforms are implicated in redox-state modulation of glia and neuronal cells.58, 88–90 They belong to the protein family of flavin-containing amine oxidoreductases and are located in the outer mitochondrial membrane in most cell types, including neurons.58, 88, 91 MAO-oxidizing activity requires the cofactor flavin adenine dinucleotide, which binds to the cysteine residue of the complex. The enzymatic reaction of MAOs uses molecular oxygen to remove an amine group from a molecule to produce the corresponding aldehyde and ammonia, by catalyzing the oxidative deamination of monoamines. In this process, H2O2 is produced as a subproduct of the reaction.58, 92
MAO-A and MAO-B are fundamental for the inactivation of monoaminergic neurotransmitters, a pharmacological target of several drugs, designed to raise levels of MAO in the CNS. In humans, MAO-A is highly expressed in the brain and liver, whereas MAO-B is abundant in the liver, lungs, and intestine. In the brain, MAO-A is mainly located in neurons, while MAO-B is preferentially expressed in glia and astrocytes, placing both subtypes of that enzyme in a position to interfere with ROS production in the CNS. 91 In neurons, MAO-A is mainly found in all catecholaminergic neurons and MAO-B is principally expressed in serotonergic neurons and glial cells. 88 MAO-A oxidizes noradrenaline and serotonin, whereas MAO-B mainly beta-phenylethylamine.
MAO enzymes are involved in the homeostasis of monoaminergic neurotransmitters. MAO deficiency leads to excessive levels of serotonin, norepinephrine, and dopamine. Large amounts of evidence demonstrate that learning and memory require controlled levels of catecholamines in brain tissues.93–97
NADPH oxidase
NADPH oxidase is a membrane-bound enzymatic complex producing superoxide through the oxidation of NADPH. As an ROS generator, the complex has a peculiarity when compared to other cell sources: while the production of ROS by mitochondria is a side product of the respiratory process, superoxide generation via NADPH oxidase is the main function of this enzyme. NADPH oxidase was first characterized as an enzyme acting in the immune system. Specifically, it is located in the plasma membrane or phagosomes of polymorphonuclear neutrophils. 98 Under normal circumstances, the complex is latent, but it is promptly activated to assemble in the membranes during respiratory bursts. Neutrophils activate NADPH oxidase to function in host defense. Superoxide is produced to kill bacteria and fungi ingested inside the phagosomes.98, 99
NADPH oxidase is made up of six subunits. One of these is a Rho protein with GTPase activity. Two sub-units (gp91phox and p22phox) are anchored to the plasma membrane, and the other three components are found in the cytoplasm (p40phox, p47phox, and p67phox).98, 99 When specific cellular events recruit the involvement of NADPH oxidase, the p47phox subunit ensures that cytosolic subunits correctly assemble the membrane components to form the NADPH complex. Once formed, the enzymatic complex catalyzes the oxidation of NADPH to NADPH+, passing an electron to molecular oxygen and forming the superoxide anion.99, 100 The protein Rho and p67phox subunit activate the gp91phox subunit, which is responsible for catalyzing the formation of superoxide. The activity of NADPH oxidase is regulated by many intracellular signaling pathways. The most crucial factor leading to the activation of the enzyme is the presence of Ca2+ in the cytosol. 101
Over the past decade, researchers have found evidence that NADPH oxidase is present in other cell types involved in nonphagocytic activities, including regulation of cellular growth and death, cellular endothelial function, and mediation of intracellular signaling.102, 103 More recent studies identified the presence of NADPH oxidase in the postsynaptic terminals of several structures in the nervous system, suggesting a possible involvement of NADPH oxidase in neuronal activity.104–107 Immunohistochemical studies in mouse and rat tissue extensively identified the distribution of NADPH oxidase in the brain, with higher concentrations in the cortex and hippocampus.105, 106 NADPH oxidase is present in cell bodies and dendrites of hippocampal neurons and colocalizes at synaptic sites with synaptoneurosomes and synaptophysin. 107
NADPH oxidase is thought to be the major source of superoxide production in the nervous system during physiological conditions. Given the importance of calcium influx for the activation of the complex, the postsynaptic localization of NADPH oxidase in neurons suggests an important role in cellular mechanisms involved in synaptic plasticity. In this context, NADPH oxidase as a source of ROS has recently attracted notable interest, since this complex is promptly activated after stimulation of the NMDA receptor.108, 109
Physiological Effects of ROS
Involvement of ROS in cellular signaling
Oxidative stress is often associated with age-dependent loss of synaptic plasticity and cognitive functions. Nevertheless, when antioxidative defenses are efficient enough to neutralize the harmful effect of oxidizing molecules (Fig. 1, point 11), the presence of ROS is fundamental for many physiological cellular processes. Several studies provide evidence that ROS participate as signaling molecules in a wide range of cellular functions. In a variety of biological mechanisms, ROS modulate intracellular transduction pathways and transcriptional factors involved in cell proliferation, differentiation, and maturation.8, 10, 110–117
Physiological Effects of Ros on Synaptic Plasticity. Nmda Receptor Stimulation (
Compared to other cellular mechanisms, redox signaling uses molecules with greater potential for nonspecific reactions. ROS have many more potential targets compared to other molecules such as cAMP.
118
ROS do not act as messaging molecule-binding receptors in the traditional sense but rather oxidizing specific amino acid residues. For example, ROS regulate cellular activities by acting on redox-sensitive cysteine residues, which are typically located in active sites and catalytic domains of protein tyrosine phosphatases.119, 120 Cysteine residues are particularly susceptible to oxidation due to low p
In the nervous system, ROS production regulates neuronal development from neuronal precursors.125–127 Redox signaling is required for cell expansion in their niches of proliferation. 128 ROS and oxidative states influence signaling cascades important for neurogenesis by modulating the redox state of tyrosine-phosphorylated proteins such as PKC and by regulating redox sensitive transcriptional factors such as the nuclear factor ºB (NF-ºB), the activator protein 1 (AP-1), and nuclear factor of activated T-cells. 129
Redox signaling is also required to trigger neuronal differentiation and axon formation.130, 131 Differentiation from neuronal progenitors to neurons is regulated through interactions involving
In cortical neurons, the pairing of H2O2 application and depolarization enhances the intracellular Ca2+ signaling, promoting a form of activity-dependent modulation of cellular excitability. 137 The temporal pairing of depolarization and oxidation is critical for this potentiation, suggesting a physiological link between functional activity and the metabolic state in neurons. In hippocampal slices, ERK phosphorylation increases after H2O2 application, and this effect is blocked by antioxidant application. 138 Likewise, exogenous H2O2 increases ERK and CREB phosphorylation in PC12 cells and cortical neurons.7, 12, 139–141
Based on these studies, many neurobiologists are investigating the possibility that ROS can act as messengers in the transduction pathways important for synaptic plasticity in the CNS.
ROS production is required for synaptic plasticity
Direct evidence shows that ROS participate in synaptic plasticity processes as second messengers in several areas of the nervous system, including the hippocampus, cerebral cortex, spinal cord, hypothalamus, and amygdala.5, 6, 9, 142–148
ROS production is necessary for hippocampal LTP, which is a form of synaptic enhancement involved in certain types of mammalian learning and memory.56, 149–156 It is generally accepted that the hippocampus participates in the processes of information storage adopting synaptic potentiation mechanisms, which are based on changes in calcium levels of dendritic spine and local protein synthesis.4, 157–159 In the CA1 area, most forms of LTP induced by HFS are dependent on Ca2+ entry into the postsynaptic neuron through NMDA receptor activation.160, 161 Genetic or pharmacological manipulations that reduce the production of ROS negatively affect the LTP, suggesting that hippocampal plasticity involved in memory formation and/or consolidation requires specific redox states.56, 149, 152, 162–164
In the amygdala, ROS mediate pain neuroplasticity by increasing excitatory neurotransmission and excitability of the central nucleus of the amygdala (CeA), which is responsible for the emotional-affective aspect of pain modulation and pain related behavior.143, 151, 165–169 Excitability and synaptic transmission of the CeA are increased in acute and chronic inflammation.165, 168, 169 Experimentally induced inflammation enhances CeA neuronal activity together with animal behavioral responses, and such effects are inhibited when SOD is administrated.143, 170, 171 ROS contribute to changes in the processing of the visceral pain response by enhancing the excitatory drive of output neurons from the amygdala, suggesting that oxidative cellular states are important for the neural processing of emotional-affective aspects of pain.143, 171
In the spinal cord, ROS act as a signaling molecule in neuroplasticity processes related to persistent neuropathic and inflammatory pain.172–174 Spinal cord LTP is thought to be the physiological substrate for sustained central sensitization, the main mechanism underlying these forms of pain.171, 175, 176 Experimental evidence suggests that ROS operate in the spinal cord circuitry during sensitization.177, 178 Application of ROS scavengers impairs the induction and maintenance of LTP in spinal cord tissue preparations. Administration of ROS donors is sufficient to induce LTP, but if HFS is applied after the establishment of ROS-induced potentiation, the LTP is attenuated. Similar effects are observed when application of HSF occurs first and the ROS donor is administrated subsequently, suggesting that overproduction of ROS is detrimental to LTP. Sensitization requires HFS to activate NMDA receptors leading to production of ROS, which in turn participate in the expression of LTP. The authors suggest that this process may be the basis of amyloid β (Aβ)-fiber-mediated allodynia. 173
The effects of ROS on synaptic plasticity were also studied through invertebrate experimental models. In
ROS and plasticity: effects of superoxide and hydrogen peroxide
Normally, ROS production during neuronal activity begins with the formation of the superoxide anion, mainly through the activity of mitochondria and NADPH oxidase. Once produced, superoxide can potentially react with nearby cellular components, but it is rapidly converted into H2O2, which is a more stable molecule than superoxide. Despite H2O2 being less reactive, its higher stability grants it a larger diffusion. H2O2 is capable of passing the cell membrane through aquaporin homologs and acts as a second-messenger molecule in nearby synaptic terminals and neurons. 181 Depending on the concentration of iron cation (Fe2+), present in molecules such as hemoglobin, H2O2 can be converted into hydroxyl radical, which is a more reactive species. 182 However, as most reactive ROS are quickly transformed into more stable molecules, distinguishing between the differential and relative contribution of superoxide, H2O2, and hydroxyl radical in the modulation of synaptic plasticity is extremely difficult.
Electrophysiological studies found that superoxide accumulates in hippocampal slices after heavy NMDA receptor stimulation. 183 Administration of exogenous superoxide scavengers blocks the induction of LTP in the CA1 area of hippocampal slices, suggesting that the presence of superoxide is necessary for synaptic LTP.149, 184 On the other hand, superoxide scavengers do not have effects on short-term plasticity or post-tetanic potentiation, suggesting that superoxide acts specifically on LTP. 149
In the hippocampus, the relationship between synaptic plasticity and superoxide production has been extensively investigated using enzymes called SOD, through either exogenous application of SOD or using transgenic animals overexpressing endogenous SOD. These enzymes and other artificial superoxide scavengers block the HFS-induced LTP in the CA1 area. 149 Exogenous administration of superoxide through xanthine = xanthine oxidase (X = XO) results in transient reduction in postsynaptic response, followed by a late form of LTP, which can be inhibited by SOD application. 152 Superoxide-induced LTP occludes HFS-induced LTP, indicating that these two forms of synaptic potentiation are based on similar mechanisms.
Neurons have three isoforms of endogenous SOD: cytosolic SOD (SOD1), extracellular SOD (EC-SOD), and mitochondrial SOD (SOD2). Mice overexpressing EC-SOD or SOD1 exhibit deficits in LTP, but the mechanisms underlying these effects are different depending on the overexpressed SOD isoform. In EC-SOD transgenic mice, the LTP is impaired because superoxide production available in the hippocampal slice is not sufficient to allow the induction of LTP.150, 185 On the other hand, SOD1 transgenic mice exhibit deficits in LTP due to the increase in H2O2 accumulation. 185 Mice overexpressing SOD2 show normal LTP, suggesting that mitochondrial ROS are unlikely to play a role in promoting synaptic plasticity. 186
The production of superoxide seems to be associated with forms of plasticity that require the abundant presence of intracellular calcium. First, as described above, superoxide is fundamental to induce LTP in the hippocampus, which requires high concentrations of intracellular calcium. Second, superoxide is important to induce LTD in the cerebellum, where synaptic depression requires a high influx of calcium. In fact, reducing superoxide availability suppresses the induction of LTD in the cerebellar Purkinje neurons. 148
In studies where superoxide was found to be required for LTP, exogenous SOD produced increased concentrations of H2O2, attenuating LTP. These results indicate that overproduction of H2O2 inhibits synaptic potentiation. 56 Also, some studies found that H2O2 impairs LTP in rat hippocampal slices187, 188: H2O2 administration (20-100 µM) facilitates LTD and suppresses several types of LTP induction, namely, synaptic potentiation based on muscarinic receptor activation, tetanic stimulation, and voltage-gated calcium channel opening. 189 However, lower concentrations of H2O2 (1 µM) cause a huge increase in tetanic LTP and enhance NMDA receptor-independent LTP. 187 Potentiation of synaptic responses is attenuated by the application of the H2O2 scavenger catalase. 150 Thus, facilitating or impairing the effects of H2O2 on plasticity seems to be dose-dependent effects.
Ultimately, superoxide production appears to be the mandatory process required to begin the redox changes that modulate plasticity. The short-living superoxide is then converted to longer lasting H2O2, which regulates synaptic plasticity in a dose-dependent manner. 187
ROS sources and physiological effects on synaptic plasticity signaling
As described in “Synaptic plasticity” section, ROS can be produced by various cellular structures. Some studies investigated the involvement of ROS in synaptic plasticity, focusing on the role of specific endogenous cellular sources. These finding are discussed below in this section.
Mitochondria
Some studies provide indirect evidence that mitochondrial ROS production might be involved in synaptic plasticity.190–192 The localization of mitochondria in dendrites is activity dependent, and intense stimuli causing high calcium influx lead to increased superoxide produced by mitochondria (Fig. 2, points 2–8).193–195 Mitochondrial superoxide release upregulates the activation of CaMKII and PKA, two kinase proteins involved in cellular mechanisms of synaptic potentiation. On the other hand, the phosphatase PP1 associated with LTD is downregulated by mitochondrial ROS.190, 191 The application of complex I inhibitors reduces the production of H2O2, the influx of calcium, and the phosphorylation of ERK.
196
The mitochondrial effects on intracellular signaling are mediated through regulation of ROS, which is influenced by activity-dependent regulation of mitochondrial motility and localization.
192
Pathological Effects of Ros on Synaptic Plasticity. Glutamatergic Over Stimulation and Aβ Oligopeptides Sustain Nmda Receptor Hyperactivation (
One study tried to directly associate the production of mitochondrial ROS with synaptic plasticity but obtained negative results in this sense. 186 The authors found that transgenic mice overexpressing SOD2 do not show any deficit in hippocampal LTP or memory. However, supporting evidence has shown that generation of ROS by mitochondria following mitochondrial Ca2+ uptake (MCU) is a key step for the induction of LTP in the spinal cord, since inhibition of MCU blocks potentiation despite the increase in cytosolic Ca2+ levels produced after NMDA receptor activation.177, 197 ROS derived from mitochondria, mainly superoxide, activate downstream signaling cascades involving PKA, PKC, and ERK. Activation of these protein kinases by superoxide is essential for sensitization at the spinal dorsal horn. 197
Neural nitric oxide synthase
ROS production through nNOS may contribute in the modulation of synaptic plasticity. However, the role that nNOS-derived ROS play in plasticity is difficult to assess. The available pharmacological approaches do not allow the selective blocking of ROS production without interfering with the generation of NO. Since ROS production becomes significant only in conditions of L-arginine deficiency, nNOS contribution in plasticity and memory is typically attributed to NO. 72 Nevertheless, one study found that nNOS expression causes an increase in ERK activation, and this effect is inhibited by the presence of SOD, suggesting that phosphorylation of ERK might occur through the production of ROS. 198 However, it is important to consider that nNOS stimulation has potentially aversive properties for the induction of plasticity. First, NO produced by nNOS may inhibit the functionality of the NMDA receptor so as to reduce the flow of calcium in the postsynaptic neuron.82, 84–87 Second, NO can be converted into peroxynitrite by reacting with the superoxide provided by other sources of ROS. In doing so, nNOS reduces the amount of superoxide available to be converted into hydrogen peroxide, and thus, to be used in other signal transduction pathways related to plasticity.
Monoamine oxidase
Studies that tried to correlate the MAO activity with neuronal plasticity and memory have adopted transgenic animals and pharmacological inhibitors. Knockout mice for MAO exhibit an increase in hippocampal LTP, which is related to changes in the expression of NMDA glutamate receptor subunits. 93 In the barrel cortex, MAO knockout mice exhibit potentiated thalamocortical excitation and feedforward inhibition. 199 However, these results are difficult to interpret with regard to the relationship between MAO and ROS production. The effects caused by MAO blockade are better attributable to the direct action over the monoamine turnover, rather than to changes in redox cellular environment.
NADPH oxidase
Both in the hippocampus and visual cortex, NADPH oxidase produces bursts of superoxide as a response to Ca2+ influx through NMDA receptor stimulation.109, 142 The NMDA-induced increase in ROS mediated by NADPH oxidase requires the activation of nNOS and protein kinase G. 200 Considering its localization in neural structures and connection with the NMDA receptor, it has been suggested that NADPH oxidase could be the major source of ROS playing a physiological role in the mechanisms of synaptic plasticity. 109 This idea is supported by the findings that human patients with mutations in genes encoding NADPH oxidase display mild cognitive deficits. 201
Activation of NMDA receptors stimulates NADPH oxidase to generate the superoxide anion (Fig. 1, point 1), which readily dismutates into H2O2. Once produced, H2O2 is capable of passing the plasma membrane to interact with pre-and postsynaptic mechanisms and affect synaptic transmission. Since NADPH oxidase produces large amounts of superoxide in a controlled manner, it appears to be the ideal source of ROS to provide rapid and controlled oxidative responses to specific stimuli of extracellular signaling. 202 One of the most important processes in this sense is the activation of ERK via NMDA receptor stimulation (Fig. 1, point 4). Inhibition of NADPH oxidase by diphenylene iodonium blocks the NMDA receptor-dependent phosphorylation of ERK in the CA1 area of the hippocampus. Mutant mice lacking the subunit p47phox, one of the essential subunits of NADPH oxidase, also show impaired LTP. 108 ROS production through NADPH oxidase participates in the intracellular pathway linking stimulation of NMDA receptors to ERK activation. Pharmacological and genetic manipulations leading to NADPH oxidase blockage abolish ERK activation induced by NMDA receptor activation. 108
Experimental evidence from animal models showed that ROS production through NADPH oxidase is primarily a physiological process important for synaptic plasticity mechanisms operating in the hippocampus and visual cortex.142, 162 Knockout animals for the subunits p47phox and gp91phox exhibit impaired LTP and reduced post-tetanic facilitation in the hippocampus, along with mild deficits in spatial memory. 162 Experiments performed in the visual cortex of gp91phox knockout mice showed that NADPH oxidase is necessary for the induction of LTP and LTD, both in young and adult animals. 142 However, applications of the NADPH oxidase inhibitors block LTP but not LTD. This suggests that visual cortical LTP requires acute bursts of ROS to be produced during NMDA stimulation, while chronic and milder activation of NADPH oxidase is sufficient for LTD mechanism to occur.
Interestingly, NMDA receptor-mediated postsynaptic responses in the visual cortex are reduced in gp91phox knockout mice. This suggests that ROS produced by NADPH oxidase might act as a positive feedback mechanism, increasing the efficacy of the NMDA receptor. Indeed, NMDA receptor function is modulated by the redox cellular environment. Some reductant species inhibit, while others enhance the activity of the NMDA receptor. 203 Oxidation abolishes the capability of reductants to modify the NMDA receptor function. ROS produced via NADPH oxidase could be directly involved in NMDA receptor redox modulation. Alternatively, NADPH oxidase-derived superoxide might influenciate NMDA receptor activity by interacting with NO. Superoxide easily reacts with NO, which is known to be a potent inhibitor of the NMDA receptor. 84 In mesangial cells, NO bioavailability is reduced by NADPH oxidase, whose activity increases the interaction between NO and superoxide to form peroxynitrite. 204 A similar mechanism could operate in neurons and could be responsible for reducing the NO-mediated inhibition of NMDA receptor. During the induction of synaptic plasticity, superoxide generated by NADPH oxidase might counteract the NO-dependent inhibition over the NMDA receptor and consequently facilitate plasticity.
The activation of NADPH oxidase is important for some processes of neuroplasticity occurring as a reaction to lesions and external insults. For example, NADPH oxidase-generated ROS drives the structural neuronal remodeling occurring after ocular enucleation. Neurons of superior colliculus and dorsal lateral geniculate nucleus react to ocular enucleation undertaking structural reorganization through increase in the production of neurofilament and microtubule-associated protein-2 (NMAP2). These effects are accompanied by elevated ROS production via NADPH oxidase, as both ROS generation and NMAP2 synthesis are inhibited by NADPH oxidase inhibitors. 205
NADPH oxidase is located at membrane sites of phrenic motoneurons of the C4 ventral horn. NADPH oxidase-derived ROS participate in respiratory plasticity of phrenic motor neurons, which is recorded in the phrenic nerve following acute intermittent hypoxia.104, 146 pLTF requires the production of ROS to stimulate protein kinases and inhibit protein phosphatases. 206 Superoxide generation is fundamental for such a process, since pLTF is blocked after application of a SOD mimetic into the cervical spinal region that contains the phrenic motor neurons. 145 Results showed that the main source of ROS involved in pLTF is NADPH oxidase, as pharmacological inhibition of this complex attenuates the expression of pLTF in a dose-dependent manner. Increased ROS formation during acute intermittent hypoxia facilitates pLTF by inhibiting specific protein phosphatases, while intravenous administration of SOD mimetics blocks pLTF.145, 146, 206 Intrathecal injections of NADPH oxidase inhibitors also attenuates pLTF, demonstrating that spinal NADPH oxidase activity is a necessary source of ROS for this form of respiratory plasticity. 104
Interestingly, when NADPH oxidase-mediated ROS are neutralized through SOD overexpression, compensatory mechanisms activate to maintain synaptic facilitation. Transgenic animals overexpressing a mutated form of SOD1 (SOD1G93A) are typically adopted as a model for ALS neurodegenerative disease. Although SOD1-elevated expression is potentially capable of decreasing oxidative stress, it promotes neurodegeneration on motor neurons by enhancing the formation of protein aggregates.207, 208 Despite superoxide accumulation being decreased in SOD1G93A animals, pLTF is enhanced at the end stage of the disease, suggesting that compensatory mechanisms maintain ROS homeostasis and preserve hypoxia-induced respiratory plasticity. 209
In the paraventricular nucleus of the hypothalamus (PVN), NADPH oxidase participates in mechanisms increasing neuronal excitability during autonomic and neuroendocrine homeostatic responses. When blood pressure decreases, Ang-II stimulates PVN neurons, whose activity increases sympathetic nerve activity to counterbalance the pressure drop. Enhanced synaptic signaling from the forebrain through PVN increases arterial pressure by inducing sympathetic long-term facilitation. 210 High circulating levels of Ang-II also trigger changes in PVN synaptic transmission through modifications in NMDA receptor functioning. 211 Upregulation of ROS in the PVN contributes to increased sympathetic activity during blood pressure and adipose tissue metabolic responses.147, 212–217 Microinjection of a SOD mimetic into the PVN decreases arterial pressure and sympathetic nerve activity, suggesting that ROS signaling contributes to Ang-II effects on hypothalamic arterial pressure homeostasis. 217 During feedback attenuation of augmented sympathetic reflexes, antioxidant enzymes are overexpressed in the PVN, suggesting that elevated ROS generation is implicated in this process. 218 These studies support the idea that NADPH oxidase plays an important role in modulating synaptic plasticity of the PVN. It is possible that ROS are active in feedback mechanisms as well, since H2O2 administration was found to tonically suppress sympathetic responses to glutamate stimulation of PVN. 219
Intracellular mechanisms of ROS-dependent plasticity
Physiological Effects of ROS on Synaptic Plasticity

In the hippocampus, ROS are important for the activation of protein kinases fundamental for plasticity and memory, such as ERK, CaMKII, and PKC, during the induction of long-lasting LTP.11, 138, 164, 183, 220, 221 Plasticity onset implies NMDA receptor opening (Fig. 1, point 1) to cause superoxide increase, mainly through the activation of the NADPH oxidase (Fig. 1, points 2 and 3).56, 108, 109, 142, 200 Superoxide is required for the activation of both PKC and ERK during LTP induction.56, 222–224 Activation of ERK through NMDA receptors requires a suitable oxidative redox state, as the presence of antioxidants and superoxide scavengers block the activation of ERK obtained through NMDA administration in hippocampal slices. 108 PKC activation is an important step for the induction of LTP, as induction of synaptic potentiation needs the translocation of PKC to the plasma membrane.224, 225 It has been proposed that oxygen radicals enhance the autophosphorylation of PKC, as well as the release of glutamate, playing a role as a modulator of presynaptic efficacy. 163
In the hippocampus, ERK activation requires superoxide to be partially converted into hydrogen peroxide, as H2O2 is fundamental for ERK and CREB phosphorylation in response to NMDA receptor stimulation (Fig. 1, points 4 and 8).7, 55, 108 H2O2 participates in the intracellular signaling of hippocampal plasticity by stimulating calcium release from intracellular neuronal stores. NMDA receptor-mediated production of H2O2 is fundamental for the opening of ryanodine receptors (RyRs). 226 RyRs are calcium channels activated by previous calcium influx and participate in various signaling pathways important for synaptic plasticity. 227 The presence of H2O2 allows RyR to provide the calcium levels required for ERK and CREB phosphorylation (Fig. 1, point 7). 108 In fact, inhibiting RyR with ryanodine diminishes H2O2-induced ERK and CREB activation. H2O2 can also stimulate ERK through direct redox modifications of the Ras protein.228, 229 H2O2 elevates the mRNA levels of the early genes c-fos and egr-1, and these effects are prevented by the RyR blocker ryanodine. Thus, the establishment of long-term synaptic changes in the hippocampus requires ERK phosphorylation and CREB-mediated gene expression, and these purposes can be achieved through the ROS-dependent activation of RyR.
The participation of H2O2 in hippocampal plasticity depends in part on postsynaptic cell iron concentration. Iron cations (Fe2+) are strong pro-oxidant elements, due to their capacity to participate in one-electron reactions. The presence of iron in the intracellular fluid is not necessarily harmful for neural functioning but may be rather beneficial, given that its concentration is kept within physiological range. Iron is important for the maintenance of the appropriated redox environment for synaptic plasticity in neurons. Fe2+ catalyzes the transformation of the mild oxidant H2O2 into hydroxyl radical (OH), which is much more reactive than H2O2. 182 In cultured cortical and hippocampal neurons, activation of NMDA receptors allows the entrance of iron inside the postsynaptic cell, resulting in OH production. Stimulation of the NMDA receptor (Fig. 1, point 1) induces calcium entry and NO production through activation of nNOS (Fig. 1, point 2), allowing Fe2+ entry through NO-mediated opening of the divalent metal transporter 1 (DMT-1; Fig. 1, point 4). NO modifies a cysteine residue in the GTPase Dexras1, thus enhancing iron uptake by activating a complex made up of Dexras1, peripheral benzodiazepine receptor-associated protein 7, and DMT-1.227, 230 In this context, iron uptake provides the neuronal redox conditions necessary for the proper functioning of cellular factors involved in synaptic plasticity. 231
The presence of iron inside the cell is necessary to stimulate the opening of RyR channels induced by ROS. Iron-generated OH amplifies the calcium signaling initiated by stimulation of NMDA receptors. 232 In PC12 cells, iron application generates calcium signals through RyR and activates the ERK pathway, while iron chelators block these effects. 233 Iron promotes formation of OH, which elicits RyR-mediated calcium signals (Fig. 1, point 7) capable of facilitating the activation of the ERK pathway (Fig. 1, point 4). After NMDA receptor stimulation, the opening of RyR channels elevates postsynaptic Ca2+ influx from endoplasmic reticulum in hippocampal dendritic spines. 234 Preincubation of hippocampal neurons in culture with ryanodine blocks both NMDA receptor-mediated intracellular calcium increase and ERK phosphorylation. 233 High concentrations of ryanodine completely inhibit RyR channels, reduce CREB phosphorylation, and prevent the establishment of LTP, 235 while lower concentrations of ryanodine activate RyR channels, allowing the shift from the early to late phase of LTP. 235 Finally, NMDA receptor activation also results in the oxidation of neurogranin (Fig. 1, point 5). 236 Oxidation or phosphorylation of neurogranin allows calmodulin to activate CaMKII, contributing to the expression of plastic changes.163, 236
In summary, during the induction of plasticity in the hippocampus, NMDA receptors provide the first rise of intracellular postsynaptic calcium concentration. Second, the activation of NMDA receptors leads to H2O2 production and iron entry. Third, H2O2 and iron react to produce the high reactive radical OH. Fourth, OH interacts with the RyR channels, which amplify the calcium signaling. Finally, the total amount of calcium present in the cytosol supports the activation of several cascades fundamental for synaptic plasticity, including the ERK and the CaMKII pathways.237, 238
Interestingly, in neural structures other than the hippocampus, the effects of ROS on the induction of synaptic plasticity also involve the activation of protein kinase. ROS-mediated synaptic potentiation of the amygdala upregulates protein kinases, as combined application of ERK and PKA inhibitors completely block the excitatory effects of ROS donors on the CeA. 9 Also, the induction of spinal cord LTP during sensitization requires increased AMPA receptor phosphorylation at the PKC, PKA, and CaMKII sites, and these effects are reversed when ROS availability is reduced. 173
As an alternative to protein kinase activation, ROS can facilitate intracellular cascades related to synaptic plasticity through the inhibition of protein phosphatase. At high concentrations, H2O2 blocks the activity of tyrosine phosphatases, thus enhancing the function of tyrosine kinases.119, 239 Phosphatases regulate K+ and other ion channels, thus inhibition of these proteins may directly alter neuronal excitability and affect the occurrence of synaptic changes.240, 241
In neurons of the phrenic motor nerve, ROS inhibit the activity of phosphatases, modifying the kinase/phosphatase balance of protein involved in respiratory synaptic facilitation. Oxidation shifts the balance toward phosphorylation and facilitates the induction and maintenance of synaptic facilitation.145, 206 ROS are fundamental for the activity of several protein kinases important for pLTF, including PKC.242, 243 In this case, protein kinase activation is enhanced indirectly by ROS-induced inhibition of protein phosphatase activity. When activated by their endogenous ligand, growth factor receptors generate ROS, which in turn inhibit protein tyrosine phosphatases. 242 This process enables receptor activity, promoting cellular pathways important for plasticity. 242
ROS-mediated downregulation of phosphatase occurs for hippocampal LTP and cerebellar LTD. During the induction of potentiation in the hippocampus, superoxide inactivates
Experiments performed in
Dopamine: balancing neuroprotection and neurotoxicity
Oxidative stress is often thought to be related to pathological conditions, affecting neurons during aging and brain disease. However, reducing synaptic efficacy through oxidative burden could be part of a physiological process, which selectively weakens neural connections in a use-dependent manner. In this regard, an interesting redox hypothesis of synaptic plasticity has been formulated, suggesting that the balance between neuroprotective and neurotoxic redox agents is controlled by the level of dopamine. 251 According to this hypothesis, dopamine release would modulate the amount of oxidative stress, affecting synapses in the cerebral cortex and hippocampus.
Stimuli correlated with reward and motivation are accompanied by increased release of dopamine widely in the brain.251, 252 Positive reinforcement acts through dopaminergic transmission to modulate plasticity at glutamatergic synapses, in order to enhance the storage and processing of important information. Thus, motivational reinforcement increases dopamine release, which in turns promotes the synapses active at the time, while decreases in dopamine release lead to suppression of inactive synapses.252, 253
In brain homogenates of mitochondria and microsome preparations, catecholamines and their metabolites were found to be endogenous antioxidants with potent scavenging effects on superoxide anions and hydroxyl radicals.254, 255 For example, oxidative stress measured through B-phycoerythrin fluorescence decay is prevented by dopamine. 256 It has been suggested that synaptic plasticity could be in part mediated by the antioxidant action of dopamine on redox cellular environment.257, 258 In this sense, dopamine would act as an endogenous antioxidant in the brain, providing protection against oxidative stress in synapses that require reinforcement. 254 Dopamine would move the redox state toward a neuroprotective direction in glutamatergic synapses, favoring spine differentiation and growth, and low levels of dopamine would promote spine suppression due to the lack of antioxidant cover against oxidative stress.
Dopamine shows a significant effect in suppressing superoxide anions and hydroxyl radicals, through a direct and indirect mechanism.
255
Direct antioxidant effects are achieved when dopamine is converted to
Dopamine receptors can be endocytosed after neurotransmitter binding. This occurs more frequently when the receptor is eventually replaced because of accumulated oxidative damage in the synapse. 260 The receptor-ligand complex is endocytosed inside the postsynaptic neuron and transported to the tubulo-vesicular endosome system. Here, the receptor-ligand complex is delivered into the lumen of the endosome, where dissociation of the ligand from the receptor occurs. Both D1 and D2 receptors are endocytosed, but only D1 receptors are internalized together with the iron transporter transferrin. 261 Since the D1 receptor-dopamine complex and transferrin colocalize in the same endosome, cheatable iron and dopamine are in close contact. Together with transferrin, the D1 receptor-dopamine complex acts as a catechol-iron complex, which is a strong antioxidant capable of transforming superoxide into oxygen and hydrogen peroxide (Fig. 1, point 10). 260 Thus, the catechol-iron complex can perform several antioxidant reactions with superoxide in the cytoplasm.258, 262
When there are low antioxidant agents in the cell, catecholamines are oxidized to form o-quinones, including the neurotoxic free radicals
Pathological Effects of ROS
Oxidative stress and age-dependent decline of neural functions
Excessive production of ROS leads to oxidative stress, which is defined as the imbalance between the production of ROS and the antioxidant cellular processes. 59 ROS accumulation modifies the conformation of several proteins and causes the oxidization of cellular components such as membrane lipids, proteins, and DNA.264, 265 Oxidative stress in a brain region occurs when ROS production is greater than antioxidant defenses. Redox mechanisms are those providing a balanced interaction between oxidative reactions and anti-oxidative (reductive) defenses. ROS accumulation in biological tissues is normally controlled by antioxidant enzymes such as vitamins, SOD, catalase, and peroxidases. 266
The brain is particularly vulnerable to oxidative stress, because it consumes a large amount of oxygen, has abundant lipid content, and has little antioxidant activity compared to other organs. The major antioxidants in the brain are ascorbate, glutathione (mainly inside astrocytes), and vitamin E in the plasma membrane. It has been proposed that the age-related decline in memory and cognition could result from ROS-induced dysregulation of Ca2+-dependent cellular processes. The aging-associated pro-oxidizing shift in cellular redox status disrupts the redox-regulated Ca2+ signaling, leading to overoxidation of redox sensitive proteins. 267
Indirect evidence that ROS accumulation can be harmful to synaptic plasticity comes from studies in which the production of ROS has been related to decay of mnemonic and cognitive abilities with age. Various works show oxidative stress linked to age-dependent decline in cognitive functions.56, 149, 150, 264, 268–274 Such pathological effects of ROS have been observed both in human patients and in animal models. Behavioral deficits related to memory tasks are associated with increases in oxidative stress in aged animals.275–278 Comparisons between young and old brain tissues revealed higher levels of ROS in aged animals when compared to young animals.144, 189, 279–285 Supplements containing antioxidants are effective in reducing the age-related deficits of spatial learning performance of rats, while chronic administration of the free radical scavenger alpha-lipoic acid improves spatial memory.286–288 Results obtained from the object recognition tests showed that mice deficient in the antioxidant enzyme SOD (Sod3-/-) don't clearly distinguish between new and familiar objects. 289 In addition, Sod3-/- mice require more days of training to improve their performance in the Morris water maze test, when compared to wild mice. 290 In old animals, behavioral deficits in the fear-conditioning test are rescued through treatment with synthetic catalytic scavengers of ROS, which prevent protein oxidation and lipid peroxidation by 50%. 284 Thus, using antioxidants to reduce age-accumulated ROS levels minimizes protein oxidation and counteracts the decay of cognitive functions.
Oxidative stress in brain diseases
ROS accumulation in the brain has been associated with the onset of neurodegenerative and psychiatric diseases, whose consequence is to reduce several neuronal cellular functions, including synaptic plasticity.291–295 ROS accumulation in neurons and the resulting oxidative stress are responsible for the loss of cognitive and motor functions in several brain diseases. 296 Alzheimer's disease (AD) is one of the most common neurodegenerative pathologies in aged populations. Aβ peptides self-aggregate to form intercellular plaques. During this process, Aβ induces membrane-associated oxidative stress, which increases neuronal vulnerability to excitotoxicity. 297 By increasing ROS generation, Aβ can lead to nuclear and mitochondrial DNA damage in neurons.298, 299
ROS production is also involved in Parkinson's disease (PD) and Huntington's disease (HD), two neurodegenerative disorders commonly found in older people. PD causes cellular death in dopaminergic neurons of the substantia nigra, severely impairing neural motor control. Oxidative damage was found in the substantia nigra during PD disease progression, suggesting that ROS may play a role in the neurodegeneration of dopaminergic neurons.24, 300 HD is a genetically inherited neurodegenerative pathology, whose cellular features derive from nucleotides repetitions of the huntingtin gene. This genetic mutation enhances NMDA receptor activity and sensitizes type 1 inositol 1,4,5-trisphosphate receptors, upsetting the intracellular calcium balance.301, 302 As a consequence of disrupted calcium homeostasis, mitochondrial function is affected, resulting in impairment of the energy metabolism and generation of ROS. 303
There is also evidence linking the production of ROS with Rett syndrome (RS). RS is an X-linked brain disorder caused by mutations in the transcription factor called methyl CpG-binding protein 2 (MeCP2). This disease affects the balance between neural excitation and inhibition, causing autismlike behavior and cognitive deficits. 304 Hippocampal slices of the RS mouse model exhibit intensified levels of oxidization, a more vulnerable redox-balance, and more intense responses to oxidative challenge, suggesting that excessive ROS production might underlie the physiopathology of RS. 305
Within the last decade, many studies have pointed out that psychiatric disorders might be partially caused by oxidative stress.292, 306, 307 This hypothesis is supported by research focused on the effects of antidepressants in animal models of stress and depression. The antidepressant
Schizophrenia has been recently correlated with elevated accumulation of ROS, thought to be responsible for synaptic plasticity deficits. 311 A subtype of patients with schizophrenia develop unbalanced excitation, inhibition, and enhanced glutamatergic activity in cortical neural circuits, due to inhibition of NMDA receptor-mediated neurotransmission and a decrease in GABAergic inhibitory modulation. The result is the excessive stimulation of AMPA receptors leading to generation of ROS and cell death via apoptotic mechanisms. 312 Taken together, these findings suggest that pathological effects of ROS production are not limited to neurodegenerative disorders but rather can be extended to a broader set of neurological and psychiatric diseases.
Oxidative stress and neuronal death
Accumulation of excessive levels of ROS activates cellular responses that lead to cell death. In cortical neurons, transient exposure to high H2O2 concentrations (200 μ) results in hydroxyl radical formation and the apoptotic process. 187 ROS participation in neuronal death is closely related to excessive glutamatergic stimulation and calcium influx during sustained periods of neuronal activity. Excessive stimulation of NMDA receptors leads to massive Ca2+ influx (Fig. 2, points 1 and 2), which may activate apoptotic pathways.313, 314 Elevated ROS production causes neuronal death in neurological and psychiatric diseases as a result of glutamate excitotoxicity increase.315–317 Accumulation of extracellular glutamate and NMDA receptor over activation induces neurotoxicity mediated by H2O2 (Fig. 2, point 4) in cortical neuronal cultures. 318 Exposition to glutamate causes oxidative damage in DNA, while intracellular Ca2+ chelators and ROS scavengers prevent this insult, suggesting that oxidative stress is an important step in the progression of glutamate-mediated DNA damage. 319 High levels of glutamate induce mitochondrial Ca2+ uptake along with increasing mitochondrial respiration. This results in ROS overproduction (mainly superoxide), which is capable of triggering cell death in hippocampal and cortical neurons.315, 320–322
Neurons have cellular systems counteracting the cytotoxicity related to glutamatergic overstimulation. Specific cellular mechanisms neutralize ROS production and/or repair the glutamate-induced DNA damage. For example, the activation of NF-κB protects neurons against oxidative stress and excitotoxicity in the hippocampus.323, 324 NF-κB is activated in response to glutamate signaling, and one important gene target of NF-κB is the SOD2, a mitochondrial antioxidant enzyme that reduces ROS concentrations.323, 325 Other cellular factors, such as the base excision repair (BER) components, are active in the DNA damage repair processes (Fig. 1, point 12). BER function was found to decline with age, suggesting the idea that cellular defenses reacting to oxidative stress in the brain decrease in an age-dependent manner.326–329
Neuronal activity often implicates intracellular signaling based on the entrance of Ca2+ passing through L-VDCC or NMDA receptors. Excessive ROS production has been found to open L-VDCC pathologically, elevating the influx of Ca2+ (Fig. 2, point 5).330, 331 Hippocampal neurons react to intracellular calcium increase by producing ROS, which in turn can induce the synthesis of interleukin 1β (IL-1β; Fig. 2, point 6) and consequentially liberate arachidonic acid (AA) from the cell membrane lipids (Fig. 2, point 7). 332 AA is an important messenger that regulates neuronal excitation; it can be released by neurons after neural stimulation to modulate synaptic transmission.333, 334 However, during the onset of pathological processes, the AA cascade leads to the production of several eicosanoid products involved in the inflammatory process. 335
Many cellular processes underlying ROS-induced cell death adopt the phospholipase A2 (PLA2) pathway. PLA2s are small proteins commonly involved in neurotransmission and capable of inducing AA liberation from the plasma membrane. The PLA2-AA pathway participates in the apoptotic cascade triggered as a response to the excess of Ca2+ influx inside the cell. Besides directly triggering cell death, PLA2s may further increase ROS levels by enhancing the calcium influx through L-VDCC. In fact, ROS scavengers and antioxidants reduce Ca2+ currents derived from L-VDCC and prevent neurons from undergoing PLA2-induced neuronal cell death. 330
The presence of ROS supports the activation of the PLA2-AA pathway by promoting the phosphorylation of the protein kinase ERK (Fig. 2, point 6). 11 Once released from the plasma membrane, AA undergoes oxidative metabolism leading to production of mitochondrial ROS in the hippocampus and cerebral cortex (Fig. 2, point 8).336, 337 Alternatively, AA can stimulate the release of cytochrome c from the mitochondria, resulting in the activation of caspases proteins (Fig. 2, point 9), which initiate the intracellular signaling, leading to DNA fragmentation and neuronal death.338, 339
Pathophysiology of ROS and impairment of synaptic plasticity
There is much literature to demonstrate that oxidative stress underlies the age-dependent decline of synaptic plasticity mechanisms required for cognitive functions.56, 144, 149–151, 264, 269–274 In old animals, impairment of hippocampal LTP and memory is mediated by elevated levels of oxidative stress.185, 189, 279, 281 The overexpression of EC-SOD reduces the LTP deficits caused by age. 340 In young animals, exogenous application of hydrogen peroxide on hippocampal slices typically causes impairment of LTP;185, 189, 237 however, in the hippocampus of aged animals, addition of exogenous ROS has no effect on LTP, even when high concentrations are administrated.185, 237 In aged rats, dietary treatment with antioxidants (vitamin E, vitamin C, and α-lipoic acid) or omega-3 fatty acids reverses the age-related decay of α-tocopherol cellular levels and rescues the expression of LTP.280, 281, 341 Thus, ROS accumulation is an important factor participating in the age-dependent decay of synaptic plasticity and consequently, in the loss of cognitive functions related to aging.
Interestingly, ROS production derived from cardiovascular diseases might also have neurotoxic effects for brain plasticity. There is evidence showing that heart disease is linked to cognitive deficits in humans. 342 Myocardial infarction reduces the blood flow in the brain, leading to the production of ROS. A recent work used an animal model of myocardial infarction to investigate the effects of vascular diseases on synaptic plasticity of the hippocampus. The LTP of the dentate gyrus was found to be significantly decreased in animals subjected to infarction. 343
ROS-dependent toxicity and synaptic plasticity
The idea of ROS involvement in brain pathology is supported by the fact that toxic substances that increase oxidative stress have negative effects on plasticity and memory. Melamine toxicity breaks down redox balance and oxidation/antioxidation homeostasis, inducing deficits in learning and spatial memory.344–346 Rapamycin can be used as an autophagy activator, reducing the melamine-induced excessive generation of ROS. Rats treated with melamine exhibit undermined LTP, LTD, and spatial learning, while rapamycin significantly rescues synaptic plasticity and the impairment of cognitive functions. 347
Nasal administration of silver nanoparticles increases superoxide and hydroxyl radicals in the hippocampus of rats. The animals also exhibit deficient LTP in the dentate gyrus and reduced spatial memory. The effects induced by nanoparticles are likely mediated by ROS, since immunohistochemical assays revealed oxidative damage in the hippocampal slices of treated animals. 348
Diets enriched with methionine result in a high plasma level of homocysteine, which causes oxidative stress. Rats fed with high concentrations of methionine showed impaired LTP in the dentate gyrus, along with deficits in locomotor skills and increased anxiety behavior. Impairment in locomotor skills is caused by overproduction of H2O2 through hyperactivation of SOD, as SOD activity was found to be elevated in the cortex of methionine-treated rats but not in the hippocampus. 349
ROS sources and pathological effects on synaptic plasticity
Main Sources of ROS and their Effects on Synaptic Plasticity
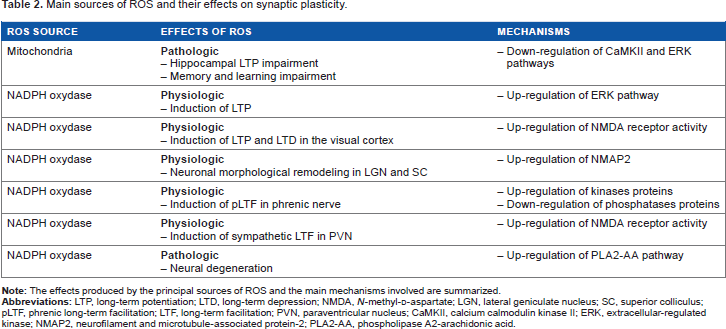
Mitochondria
The pathological consequences of mitochondrial ROS generation have been observed in a larger quantity of published studies, compared to the physiological effects. Mice lacking the intracellular ROS scavenger peroxiredoxin II (PrxII-/-) show more prominent age-dependent mitochondrial decay and LTP decline in the hippocampal CA1 pyramidal neurons, together with learning and memory impairments. 350 PrxII-/-mice fail to activate synaptic plasticity-related cellular signaling pathways involving CREB, CaMKII, and ERK. In aged PrxII-/-mice, vitamin E reduces cognitive decline by partially compensating deficits related to peroxiredoxin II deficiency, including the rescue of CREB intracellular signaling. The RS mouse model methyl-CpG-binding protein 2-deficient mouse (Mecp2-/y) is more vulnerable to cellular redox balance, due to metabolic alterations in mitochondrial function resulting in increased oxidative stress and LTP deficits.304, 305, 351 Incubation of Mecp2-/y hippocampal slices with the vitamin E derivative ROS scavenger damps neuronal hyperexcitability, improves synaptic short-term plasticity, and restores LTP. 351 The Angelman syndrome mouse model exhibits high levels of superoxide in the hippocampus, and these levels can be reduced after application of a mitochondria-specific antioxidant. In these animals, the antioxidant rescues the neurodevelopmental impairments of hippocampal synaptic plasticity and memory deficits.
In the CeA of amygdala, activation of metabotropic glutamatergic receptors type 5 produces intracellular superoxide from mitochondria. Superoxide stimulates ERK and PKA to increase neuronal excitability. 9 This ROS-mediated increase in excitability reflects in an augmented nocifensive response (response to pain or discomfort) and affective behavior and may be the mechanism underlying disorders without or with little tissue injury detected, such as irritable bowel syndrome and fibromyalgia.
In the
Taken together, these findings suggest that mitochondrial-derived ROS are more likely to exert pathological effects on synaptic plasticity, memory, and cognition, rather than physiological roles in neural cellular signaling.
Monoamine oxidase
Some experimental evidences suggest that ROS production via MAOs might contribute to oxidative stress. Aged animals subjected to a procedure inducing PD neurodegeneration were treated with a drug inhibiting the MAO activity. This treatment significantly attenuated the loss of striatal dopamine transmission. The same drug improved synaptic plasticity in the hippocampus and cognitive deficits. 353 However, such effects caused by MAO blockade may be caused by changes in the catecholamine-mediated neuromodulation rather than to changes in ROS accumulation.
The MAO inhibitor
NAPH oxidase
Some studies highlighted that NADPH oxidase could be involved in the brain pathologic consequences of ROS. There is evidence showing that modification of the activity of this complex might be linked to alterations in neuroplasticity and cognitive functions. For example, in microglial CR3 cells of the hippocampus, NADPH oxidase suppresses neuronal synaptic transmission in response to traumatic events. 359 Hypoxia and inflammatory stimuli can act synergistically to strongly activate NADPH oxidase. Once hyperactivated, the complex produces superoxide, which promotes AMPA receptor internalization in nearby postsynaptic terminals, eliciting LTD. This type of LTD may contribute to synaptic weakening and memory deficits occurring in neuroinflammation-related brain disorders. Thus, glial NADPH oxidase deregulates plasticity in the direction of LTD, contributing to pathological conditions of neuronal functioning.
The activation of NADPH oxidase can participate in the intracellular signaling, facilitating the onset of neurodegenerative diseases. Increased expression of NADPH oxidase subunits has been observed in Alzheimer diseased brains. 270 Oligopeptides Aβ interact with the NMDA receptors and this mechanism requires ROS through the activation of NADPH oxidase. Excitotoxic effects of Aβ are inhibited by NMDA receptor antagonists such as memantine, a drug commonly used to treat AD patients, suggesting that excessive NMDA receptor and NADPH oxidase activation are key events for the intracellular pathways leading to neural degeneration. 360 The NMDA-induced activation of NADPH oxidase triggers signaling pathways, leading to ERK phosphorylation. 109 ERK activates the calcium-dependent PLA2 (cPLA2), which is involved in inflammatory and apoptotic signaling. ERK cascade activation through Aβ peptides requires the NADPH oxidase-dependent redox signaling, as Aβ-induced phosphorylation of ERK is blocked by inhibitors of NADPH oxidase. 11 Thus, ROS generated by NADPH oxidase activate ERK, which in turn activates cPLA2. In addition, Aβ-induced ROS production leads to AA release from the cell membrane. Aβ-induced AA release is inhibited by NMDA receptor antagonists, suggesting the action of Aβ through the NMDA receptor. 360
Mechanism of ROS physiopathology in synaptic plasticity
Pathological Effects of ROS on Synaptic Plasticity.

ROS overproduction suppresses hippocampal LTP, interacting with different cellular pathways. For example, exposure to H2O2 undermines the expression of the NMDA-independent LTP induced through muscarinic receptor activation or the voltage-gated calcium channel. 189 H2O2-mediated suppression of LTP involves the activation of phosphatases calcineurin and PP1 (Fig. 2, point 10). This same pathway facilitates the occurrence of LTD.144, 245 LTP occlusion mechanisms include the activation of PP2A, the release of AA through IL-1β and the blocking of the phosphoinositide 3-kinase/Akt pathway via activation of glycogen synthase kinase 3β.4, 279, 343, 361 In the hippocampus of aged rats, LTP deficits are compensated with dietary supplementation including AA and its precursor γ-linolenic acid. Together, AA and γ-linolenic acid restore AA concentration to levels observed in young rats. 362 Administration of ketone bodies lowers intracellular levels of ROS and prevents the impairment of hippocampal LTP caused by H2O2-induced activation of PP2A. 361 Thus, the oxidative blockage of LTP seems to be mainly associated with the activation of the AA and PP2A pathways, and antioxidants counteract this effect by reducing ROS concentrations. Some cellular pathways activated during ROS-mediated inhibition of LTP are the same cascades potentially capable of inducing apoptosis in neurons. This suggests that cellular death might occur as a final long-term result of physiopathological pathways that begin with the suppression of synaptic connections.
ROS can inhibit hippocampal neuroplasticity through direct action on the NMDA receptor. Age-related changes in intracellular redox state contribute to the decline of NMDAR function through CaMKII (Fig. 2, point 11). The enhancement of the NMDA receptor responses depends on CaMKII activity, as this effect is blocked by the presence of CaMKII inhibitors.
363
Alterations in the endogenous reducing agent
In the cerebellum, oxidation inhibits signaling pathways essential for the NO-dependent LTP at the parallel fiber-Purkinje cell synapses. This form of LTP is impaired in aged mice. It has been proposed that such impairment might be caused by ROS-induced protein oxidation. In cerebellar slices from young mice, LTP at Purkinje cells is blocked after incubation with oxidizing agents or thiol blocker.
365
ROS blocks the LTP through the inhibition of the NO-induced protein
Conclusions
This review provides evidence that the effects of ROS on synaptic plasticity are ambivalent. The production of ROS during normal signaling is essential for the establishment of plastic changes, while excessive ROS production and consequent ROS accumulation lead to detrimental processes that impair synaptic plasticity and cause cellular injury. Oxidative stress occurs when antioxidant defenses cannot balance the production of ROS. The age-dependent decline in synaptic plasticity and memory is closely related to oxidative stress. ROS overproduction is involved in several neurologic and psychiatric pathologies, including AD, PD, depression, and schizophrenia.
The production of ROS begins with the formation of superoxide from oxygen. This first stage is essential for plasticity, given that superoxide scavengers prevent the induction of LTP.149, 152, 184 The main sources of superoxide generation are mitochondria and NADPH oxidase. Investigations into a possible direct link between mitochondrial ROS and LTP presented negative results. 186 This could depend on the limited diffusion of mitochondrial superoxide across the two membrane barriers. On the other hand, the importance of NADPH oxidase for the induction of plasticity has been reported for the hippocampus, PVN, spinal cord, thalamus, and cerebral cortex (Table 2).104, 108, 142, 162, 205, 212 In the physiological context, NADPH oxidase seems to be the major source of ROS during synaptic plasticity, and evidence suggests that NADPH oxidase-produced ROS participate in the activation of intracellular transduction pathways required for synaptic changes. This is supported by the fact that the main function of NADPH oxidase is producing superoxide in dendritic spines as a response to specific extracellular signaling. Thus, the redox signaling required for plasticity is likely to be associated with NADPH oxidase, which provides bursts of superoxide during NMDA receptor stimulation.
NADPH oxidase generates superoxide in the extracellular space. Superoxide itself might directly affect the induction of plasticity, for example, by interacting with the extracellular components of the NMDA receptor or by reacting with NO. However, superoxide is rapidly dismuted into hydrogen peroxide, which is a more stable molecule and can diffuse inside the neuron, modifying the entire cellular redox environment. In fact, H2O2 participates in the recruitment of important protein kinases (PKC, CaMKII, and ERK) whose activities are known to be critical for the establishment of long-term synaptic modifications.7, 12, 129, 139–141
Intracellular signaling underlying synaptic plasticity appears to rely on mild and controlled levels of H2O2 in the cytoplasm. In the hippocampus, a moderate increase in H2O2 supports synaptic potentiation, while higher concentrations decrease LTP, facilitate LTD, and lead to cell death. 187 The activation of pathways responsible for synaptic loss and apoptosis seems to depend significantly on H2O2 concentration levels. Intracellular transduction is probably organized to react to high levels of hydrogen peroxide by stimulating the PLA2-AA cascade. Once this pathway is stimulated, the synapse inhibits the potentiation mechanisms and, if the oxidative stress conditions persist, eventually results in the activation of neuronal death processes. Interestingly, not all ROS intracellular modulating activities are directly attributable to H2O2. In the cytoplasm, iron cations can react with H2O2 to generate hydroxyl radical, which opens the RyR Ca2+ channels, so amplifying the initial rise in Ca2+ levels mediated by NMDA receptor activation.230–235
Research about ROS-dependent plasticity still presents unsolved issues, requiring further investigation in future studies. The majority of results obtained so far are derived from LTP experiments performed in the hippocampus. Associative cortical areas involved in cognition have not been investigated. It is still unclear whether modulatory mechanisms identified in the hippocampus can be generalized to other brain areas. Likewise, investigations into the effects of ROS on LTD have received limited space, and this should be further expanded in future research. Harmful ROS-mediated effects on plasticity might occur in different manners. All possibilities for plastic changes may be completely blocked, or alternately, one specific type of plasticity may be affected. For example, glial ROS production facilitates LTD in glutamatergic cortical neurons, causing the suppression of synaptic transmission, which might be related to cognitive deficits. 359 For this reason, more comprehensive studies should investigate how ROS affect both types of synaptic plasticity, in order to better evaluate modulatory and degenerative effects and better relate ROS activity to altered brain functions.
Although it seems clear that heavy amounts of accumulated ROS are crucial for the prevalence of pathological over physiological processes, the levels of tolerance and toxicity have been poorly characterized. Works that attempted to establish a dose-dependent relationship between ROS and plasticity have been few, and future investigations should also proceed further in this direction.
Finally, it is important to emphasize that interaction mechanisms between ROS and cellular factors also require deeper investigation. As we described, in many cases redox states change the balance between protein kinases and phosphatases, in favor of kinase activity. However, most studies assessed the resulting increase in kinase activities, without investigating the underlying processes. Therefore, it is still unclear in which cases ROS directly improves the functioning of protein kinases and in which cases they reduce the activation of protein phosphatase.
Interestingly, a research line has investigated the role of dopamine as an antioxidant, proposing an idea that overlaps the boundaries between the physiological and pathological roles of ROS.252,257,258 The catecholamine redox hypothesis suggests that selective suppression of specific synapses could take place through changes in dopaminergic neuromodulation. According to this hypothesis, reducing dopaminergic transmission would select some synapses for elimination by increasing their susceptibility to oxidative stress. Unfortunately, this line of research has not been further investigated in recent years, although this perspective might radically change the current opinion about the pathological and physiological effects of ROS on synaptic plasticity.
Author Contributions
Wrote the first draft of the manuscript: RDP and TFB. Contributed to the writing of the manuscript: JF-O. Agreed with manuscript results and conclusions: RDP, TFB, and JF-O. Jointly developed the structure and arguments for the manuscript: RDP, TFB, and JF-O. Made critical revisions and approved final version: RDP, TFB, and JF-O. All authors reviewed and approved the final manuscript.