
Abstract
It has long been appreciated that in the visual cortex, particularly within a postnatal critical period for experience-dependent plasticity, the closure of one eye results in a shift in the responsiveness of cortical cells toward the experienced eye. While the functional aspects of this ocular dominance shift have been studied for many decades, their cortical substrates and synaptic mechanisms remain elusive. Nonetheless, it is becoming increasingly clear that ocular dominance plasticity is a complex phenomenon that appears to have an early and a late component. Early during monocular deprivation, deprived eye cortical synapses depress, while later during the deprivation open eye synapses potentiate. Here we review current literature on the cortical mechanisms of activity-dependent plasticity in the visual system during the critical period. These studies shed light on the role of activity in shaping neuronal structure and function in general and can lead to insights regarding how learning is acquired and maintained at the neuronal level during normal and pathological brain development.
For mammals with binocular vision reared in a normal visual environment, the majority of neurons in binocular visual cortex respond to stimulation of either eye, with some cells being preferentially driven by one eye. The frst report that visual cortical activity is shaped by experience came from the seminal studies of Hubel and Wiesel. They showed that kittens visually deprived since birth failed to develop mature cortical response properties and that monocular deprivation (MD) following a period of normal visual experience led to a decrease in the number of cortical neurons responding to stimulation of the deprived eye.1,2 This latter result of a shift in ocular dominance (OD) toward the non-deprived eye has been described by numerous subsequent studies (for reviews see) 3 7 and in several species. 8 10 It is also well established that this ocular dominance plasticity is only possible during an early postnatal critical period when neuronal connections are still being established and declines with age, 11 from about postnatal day (P) 21–35 in the mouse, 9 although plasticity outside of this critical developmental window is now being described in rodents. 12 14 Modification of visual experience has known consequences for visual function as well. For example, MD during the critical period results in a significant decrease in visual acuity in the deprived eye 15 and, when prolonged into adulthood, has also been found to produce a heightened visual acuity in the nondeprived eye. 16
The control of experience-dependent plasticity during the critical period has been localized to primary visual cortex (V1), although there is growing evidence that subcortical regions may play a role. Ganglion cells from each eye relay to the lateral geniculate nucleus (LGN) of the thalamus where they synapse on cells in specific LGN lamina such that retinotopy is preserved (Fig. 1a). The ganglion cells whose receptive fields lie within the central, binocular visual field decussate to the contralateral thalamus, leaving axons of monocularly driven cells, with receptive fields in the periphery, to travel on to the ipsilateral thalamus. LGN relay cells project to the ipsilateral primary visual cortex (V1) synapsing predominantly on cells in layer 4, which in turn project to cells in layer 2/3 (Fig. 1B). Both V1 and the LGN have been examined following periods of MD and while LGN cell size changes have been observed after prolonged deprivations, these changes have been shown to arise as a consequence of reduced cortical NMDA receptor activity,17,18 thus attributing the locus of activity-dependent visual cortical plasticity to the cortex. LGN networks, however, are known to be sensitive to visual activity
19
and recent work, such as the examination of retinogeniculate afferents after delayed dark rearing by Hooks and Chen20,21 and functional magnetic resonance imaging of LGN function in human amblyopes
22
describe deprivation effects in the thalamus which may contribute to cortical plasticity.
The architecture of the rodent visual system.
Fifty years after the discoveries of Hubel and Wiesel,1,2 we have yet to fully understand how the non-deprived eye comes to dominate cortical neurons that would otherwise have been driven binocularly. MD has been studied after both brief and extended time periods and shifts in ocular dominance can occur within a week,
23
a few days,9,24–26 1 day27,28 or even a few hours.
29
As implied by the term ocular “dominance”, the long-standing tenet was that OD shifts are the result of a “winner take all” competition between the two eyes’ cortical afferents in which the more active open eye inputs win.
30
32
Yet, we now know that the timecourse of OD plasticity is complex (Figs. 2A/B) and comprises two phases that separate changes in the two eyes: a depression of the physiological responses to stimulation of the deprived eye precedes enhancement of non-deprived eye responses.
24
Thus the question remains as to the mechanisms by which the two eyes’ response properties are regulated during OD plasticity. If competition is involved, what are retinal afferents competing for and how? This contest could be spatial, such that the inputs compete for postsynaptic area and/or resources such as neurotrophic factors,33,34 or temporal, driven by the timing of pre and postsynaptic activation.
35
How do such competitive mechanisms influence other non-competitive processes that are initiated by MD? Here, we will review literature that has addressed binocular competition, laminar loci of plasticity and structural and molecular changes associated with activity-dependent visual cortical plasticity.
Model of plastic changes during ocular dominance plasticity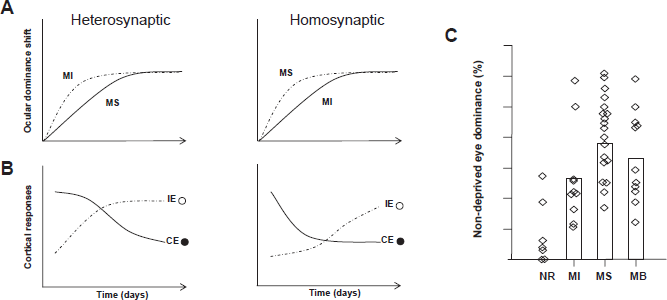
Ocular Dominance Plasticity and Binocular Competition
Modifications of synaptic strength are necessary for encoding responses to sensory experience. Predating the early MD experiments of Hubel and Wiesel, Hebb proposed that synapses strengthen when the presynaptic cell “repeatedly and persistently” excites the postsynaptic cell. 36 In 1973, Stent reasoned that the opposite of Hebb's rule was also possible, that synaptic weakening occurred when a “reduction in frequency of use of one set of synapses permits the other to take complete charge”. 37 Activity-dependent refinement of cortical response properties after MD is believed to involve long term potentiation (LTP) of non-deprived eye synapses and long term depression (LTD) of deprived eye synapses. Yet how these modes of plasticity regulate binocularity after MD and whether more global types of plasticity, such as synaptic scaling, also play a role is still unclear.
Good evidence exists that LTD of cortical synapses serving the deprived eye underlies the frst phase of OD plasticity where deprived eye responsiveness is lost, and several forms of LTD induction have been observed in visual cortex.24,38–40 Classical competition would suggest that heterosynaptic LTD would be important for OD plasticity. In this type of LTD deprived eye synapses become weaker as a consequence of the strengthening of non-deprived eye synapses on the same cortical neurons. Thus heterosynaptic LTD would be greatest at inactive synapses.37,41 Homosynaptic LTD, on the other hand, is synapse specific and requires activity at the depressing synapse.36,42–44 Homosynaptic LTD was frst observed experimentally following long lasting low frequency stimulation (1–5 hz for 5–15 min) of hippocampal CA1 neurons45,46 and can be induced in visual cortical slices following a similar protocol.
47
There is also evidence that the second phase of OD plasticity which involves a slower strengthening of non-deprived eye responses is mediated by LTP, although this phase of plasticity has not been studied as intensely. Homosynaptic LTP has been described in visual cortex53,54 and disruption of molecular processes underlying LTP also disrupt OD plasticity
55
and potentiation of non-deprived eye responses.
13
More recently, another mechanism for regulation of synaptic strength was identified that also addresses the question of what happens after the initial homosynaptic LTD of deprived eye responses. Changes in global neuronal activity elicit a form of synaptic plasticity that increases or decreases the strength of the full complement of a neuron's synapses to maintain overall stability.
56
In dissociated rat cortical cultures, blockade of activity with TTX resulted in a global multiplicative scaling up of overall synaptic strength. Scaling of AMPA receptor clustering and activation57,58 and NMDA receptor surface expression
57
have also been described. A role for synaptic scaling in the second phase of OD plasticity is supported by the paradoxical finding that monocular neurons in visual cortex that respond to the deprived eye slowly increase their responsiveness after MD.
59
This suggests that, in the absence of non-deprived eye activation, LTD is not implemented and a lack of synaptic activity instead leads to scaling up of all synapses which, in this case, are driven by the deprived eye. Additionally, the strengthening of non-deprived eye responses has been shown to be dependent on glial cytokine signaling
60
that produces synaptic scaling
Taken together, computational models and
The mode of synaptic depression induced during MD likely depends not only on the activity history but also the local cellular network. Several recent studies describe changes in cortical response properties following MD that are restricted to specific cortical laminae.
Laminar Specificity of Deprivation-Induced cortical changes
The visual cortex is a highly ordered structure. Indeed, in higher animals, cells with similar response properties are grouped together in eye specific and orientation columns. Given the existence of these local networks, it stands to reason that visual deprivation may affect distinct connections in different ways. The data reviewed above suggest that the consequences of MD cannot be explained by one single form of synaptic plasticity. Additionally several recent studies, describe lamina-specific changes in cortical response properties following MD. Thus, the mode of plasticity induced is likely to depend not only on the history of activity but also on a relationship between mechanisms in local cellular networks.
A number of studies point to extragranular layers as being the prime movers in OD plasticity. For instance, short MD (one day) results in OD shifts in layer 2/3 but not layer 4 as assayed using single unit recording in cat visual cortex.
28
This is somewhat surprising in light of the fact that layer 4 is the primary geniculate input layer and thus is the first cortical stop for incoming visual activation. However, the possibility that extragranular layers guide geniculocortical remodeling is consistent with the finding that layer 2/3 plasticity developmentally outlasts layer 4 plasticity.
69
73
Layers 2/3 remain plastic into adulthood; whereas layer 4 plasticity is lost after an early critical period in both visual69,71 and barrel cortices.74,75 Additionally, plasticity of synaptic structure occurs rapidly in layer 2/3 but not in layer 4.48,76 Homeostatic scaling also exhibits a critical period in layer 4
66
but lasts into adulthood in layers 2/3.
77
These findings, however, need to be reconciled with several other studies that show rapid OD plasticity in layer 4 using VEP
24
and single unit recordings,
78
and others that have shown plasticity of layer 4 in adulthood with titanic stimulation of geniculocortical afferents
Studies on the mechanisms of plasticity in different cortical layers support the notion that different pathways can be independently regulated during development. Early
Another important regulator of laminar plasticity may be the local networks of inhibitory neurons, as maturation of cortical inhibition is widely posited to underlie the ability to induce OD plasticity in mature cortex.
55
In cats
82
and rats
83
layer 4 lacks the horizontal inhibitory networks present in layer 2/3 (Fig. 1b) and inhibition may therefore serve as a gate that governs the induction of plasticity in different lamina and at different ages.
84
Support for this hypothesis comes from several sources, including the sensitivity of LTP in layer 2/3 following white matter stimulation
The understanding of how specific networks contribute to OD plasticity has been hampered by the fact that i
The question still remains whether extragranular plasticity is instructive for later changes in layer 4 and why the superficial layers remain plastic longer. While laminar plasticity may refect the molecular identity of neurons in different layers, it is more likely that individual synapses are regulated independently depending on the source of their input. Neurons in extragranular layers are densely interconnected through lateral, long-range connections (Fig. 1B) that are largely absent from layer 4. 82 Thus laminar differences may exist as a result of the spatial distribution of synapses carrying information from the deprived or non-deprived eye and synapses that are part of feedforward vs. short or long-range intracortical networks.
Structural Changes Associated with OD Plasticity
Changes in response properties of neurons during MD rely on adjustment of synaptic weights through potentiation and depression of the appropriate synapses. These electrophysiological modifications have been shown to correlate with profound structural changes resulting in the deprived eye losing cortical territory to the non-deprived eye following prolonged MD.10,73 This loss of cortical territory is a result of the slow retraction of geniculocortical axonal arbors serving the deprived eye and a subsequent expansion of non-deprived eye afferents. 88 90 These changes are not apparent after brief MD which elicits profound functional changes in the responses of cortical neurons to visual input through the deprived eye, suggesting that structural axonal changes are not a requirement for functional OD plasticity. Indeed brief MD does not alter the density of geniculocortical synapses serving the non-deprived and deprived eyes, 91 suggesting that more subtle changes in axonal structure do not underlie rapid functional plasticity. Thus structural changes in connectivity and cellular architecture have traditionally been thought to act as slower consolidating mechanisms that follow the rapid functional changes implemented through the alterations in molecular composition of the synapse.
Recent experiments linking postsynaptic structure to synapse function, however, suggest a possible role for fast structural alterations in dendritic spines (Figs. 3A/B) in the implementation of rapid OD shifts.
92
Dendritic spines are the postsynaptic sites of the vast majority of excitatory synapses in the visual cortex. Spines are morphologically diverse and their structure is highly dynamic.
93
95
Spine structural dynamics are activity-dependent. In slices and in culture, spines are stabilized by neuronal and synaptic activity,
96
and become more dynamic if activity is pharmacologically reduced,97,98 paralleling the developmental stabilization of spine structure as synaptic function matures.92,96 Additionally, both LTP and LTD have structural correlates at the level of the dendritic spine. In the hippocampus, induction of LTP is accompanied by rapid increases in the size of spine heads
99
101
which is closely tied to functional plasticity both in timescale and mechanism. The co-regulation of spine function and morphology is supported by the tight correlation between the expression of AMPA receptors and spine size Structural changes in dendritic spines in the visual cortex during the critical period.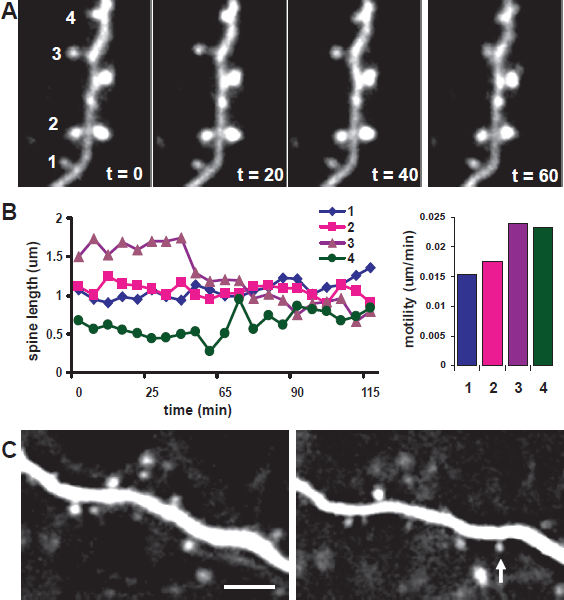
It is unclear how these structural changes may be involved in functional plasticity. One possibility is that spine size is altered by the recycling of plasma membrane during the exocytosis or endocytosis of synaptic receptors 106 which implement functional changes. Alternatively, spine structure may regulate biochemical compartmentalization at synapses and activity-driven changes in the spine head and neck can alter synaptic signaling. 107 In particular, calcium compartmentalization at the spine, which is crucial for the further implementation of plasticity (metaplasticity) is sensitive to spine structure 108 and is altered after LTP induction. 109 While these studies show that changes in spine size are linked to functional changes in synapse strength, the two phenomena can in some cases be dissociated mechanistically 110 and whether structural changes are necessary to implement plasticity remains to be shown.
Structural dynamics of axons and their presynaptic structures are even less well understood. While some studies have found axonal structures to be more stable than nearby dendritic spines,111,112 others have described high levels of structural change in axons both
Although it is well established that dendritic spine structure and density are sensitive to visual stimulation,
117
119
the progression and location of changes in dendritic spines are not well understood. The majority of studies have described reductions in spine density only after prolonged MD,
120
122
but recent technical progress allowing imaging of spine dynamics
The locus and timing of structural changes can tell us about the nature of plasticity occurring at individual synapses. Homeostatic plasticity would be expected to occur globally on all cells that experience reduced visual drive. Homosynaptic plasticity, on the other hand, would be limited to those synapses undergoing potentiation or depression and could be expressed in a very controlled spatiotemporal pattern. We know that spine motility is increased following all types of deprivation paradigms, including monocular
76
and binocular deprivation
93
as well as dark rearing;
128
thus spine destabilization may refect a compensatory mechanism activated by the loss of visual drive. It is indeed a possibility that spines enter a plastic state during loss of synaptic activity as a means to homeostatically adjust cortical activity. In fact dark adaptation in adult animals briefly re-establishes a more immature and plastic cortical state.
129
However, rapid spine destabilization by monocular deprivation was found to be limited to the binocular cortex
76
suggesting that this mechanism might be common to synaptic regulation during both brief MD and prolonged binocular deprivation. The rapid gain of spines in adult visual cortex following MD is also restricted to the binocular segment and retains many similarities to
One confound of studying the synaptic changes that underlie OD plasticity is the current inability to infer connectivity and function of individual synapses. This is unfortunate as it is readily apparent that not all synapses are equal when it comes to OD plasticity. In the adult mouse cortex, superficial spines on layer 5 neurons undergo profound and rapid structural changes following MD, whereas dendritic spines on layer 2/3 neurons do not 123 despite the fact these cells show functional changes following MD. 12 In the somatosensory cortex, anatomically distinct layer 5 neurons also show different behaviors with respect to spine remodeling following sensory deprivation, 133 and spine loss following enucleation has been described on “deep” but not “superfcial” layer 5 neurons in layer 4. 134 Additionally, different synapses may undergo plasticity at different timescales following MD. The precise wiring of the cortical circuit is still unknown and it is likely that nearby synapses serve different cortical networks and use different rules to remodel during MD. Following changes in dendritic spine structure, however, will offer valuable insight as to how OD plasticity progresses on a synapse by synapse basis.
Molecular Mechanisms
Elements of the molecular bases of activity-dependent synaptic plasticity are continually being discovered. And the evolving recognition that the mode of plasticity depends on the developmental stage and cortical lamina, as well as induction method, certainly predicts that the molecular mechanisms are just as diverse. In fact many different molecular components of OD plasticity have already been identified. These include proteins with synaptic functions such as kinases, immediate early genes, and growth factors,
135
138
as well as proteins that link calcium signaling to actin remodeling within the cell.
139
Interestingly, the extracellular environment has also been shown to strongly influence OD and synaptic plasticity.
140
Here we review studies of synaptic calcium activity and intracellular and extracellular signaling: molecular pathways thought to link synaptic activity during MD with the concomitant structural dynamics of dendritic spines (Fig. 4). We also examine what is known about plasticity beyond the critical period and how this informs our understanding of the molecular mechanism of OD plasticity.
Molecular mechanisms involved in synaptic remodeling during OD plasticity.
Synaptic Calcium Signaling
Calcium signaling at the synapse appears to be a central mechanism for initiating many types of synaptic plasticity, including OD shifts following MD. The level of intracellular calcium in the postsynapse is thought to determine the magnitude and direction of plasticity, with small calcium accumulations leading to LTD and large calcium accumulations leading to LTP.141,142 The majority of calcium entry at excitatory cortical synapses is mediated through NMDA receptors. 143 Because the magnesium blockade of NMDA receptors is released by co-activation of AMPA receptors and Na+ channels, it follows that postsynaptic calcium accumulation is activity-dependent. 144 However, even at resting potentials NMDA receptor-dependent calcium accumulations can be detected, 145 showing that even the smallest synaptic activations lead to calcium entry and can potentially trigger plasticity. NMDA receptors are also critical in OD plasticity. Local 146 148 or systemic 18 infusion of the NMDA receptor antagonists results in decreased OD shifts. Additionally, NMDA subunit composition is regulated developmentally, with NR2B subunits which have slower kinetics and result in larger calcium transients149,150 being replaced with NR2A subunits.151,152 This subunit switch is regulated by activity and dark exposure decreases the NR2A to NR2B ratio. 153 This provides a means for altering the plasticity of cortical synapses by varying synaptic calcium entry in a specific spatiotemporal pattern. Interestingly, in CA1 pyramidal neurons, synapses containing NR2A and NR2B subunits are intermingled but distinct, 154 suggesting that individual synapses in the hippocampus, but also possibly in visual cortex, can regulate their NMDA subunit composition and calcium entry profile based on their activity history. Additionally, GluR2-lacking AMPA receptors are also permeable to calcium and are expressed after LTP in the hippocampus, which may in turn promote their replacement with calcium-impermeable AMPA receptors. 155 While it is unclear whether calcium entry through AMPA receptors contributes to OD plasticity, recent evidence suggests that AMPA receptor subunit composition is sensitive to visual stimulation. 156 An additional important pathway for calcium entry into dendritic spines is through voltage-sensitive calcium channels (VSCCs) elicited by strong depolarization provided by synaptic summation and back propagating action potentials.143,144 In the cortex, calcium entry in dendritic spines is mediated by L, P/Q and low threshold T-type channels, 157 while hippocampal CA1 neuronal spines contain R and L-type channels.158,159 While the distribution of these channels between different spines on the same and different cell types in visual cortex is still not well understood, it is possible that regulation of these calcium entry pathways can provide heterogeneity in the modes of plasticity at different synapses during MD. The contribution of calcium release from internal stores after synaptic activation is more controversial144,160,161 and has not been carefully examined in the visual cortex.
All of the calcium entry pathways described above likely contribute to OD plasticity. A role for activity-dependent rises in postsynaptic calcium has been described for homosynaptic plasticity.162,163 In this scenario, action potentials that precede synaptic events elicit small calcium accumulations through NMDA receptors postsynaptic to deprived eye afferents; resulting in the depression of deprived eye responses. Supratheshold synaptic stimulation that elicits supralinear calcium entry through NMDA receptors and VSCCs postsynaptic to non-deprived eye afferents leads to potentiation of responses to the non-deprived eye. The requirement for calcium in homeostatic plasticity is less well established, although a number of studies also suggest that integrated calcium levels supported by VSCCs are responsible for the scaling of synaptic responses.164,165
Intracellular Signaling
Calcium entering the spine cytoplasm through the different receptors and channels can play various roles in plasticity by activating distinct signaling pathways. The specificity of calcium signaling is achieved through the organization of the postsynaptic density where several molecular signaling complexes associated with specific calcium entry mechanisms are brought together (Fig. 4). High resolution immuno-electron microscopy studies have revealed that the PSD is composed of three main functional layers. 166 The first contains transmembrane proteins, membrane receptors and ion channels. Glutamate receptors are a prominent part of this layer with NMDA receptors located at the center and AMPA receptors at the periphery showing the spatial organization of different signaling networks. Calcium channels, on the other hand, are located peripheral to the PSD 167 and likely associate with a different set of molecular complexes. The second layer contains scaffolding proteins that are oriented perpendicular to the PSD and which link membrane receptors with effectors through PDZ domain interactions, thus creating the dense scaffold of the PSD. The third layer contains cytoskeletal regulating proteins that are associated with the actin scaffold of the dendritic spine cytoplasm. This ultrastructural organization allows specific members of a molecular signaling pathway to be spatially grouped together such that very local changes in calcium concentrations associated with particular receptors can affect particular signaling complexes specifically. The existence of these “calcium nanodomains” has now been confirmed using calcium imaging approaches that demonstrate that L-type calcium channel flux, undetectable on the level of the spine calcium signal, activates a CaMKII and cAMP-dependent pathway and that this activation results in the depression of R-type calcium channels. 159 Additionally, small conductance Ca-activated potassium channels (SK channels) are specifically activated by calcium flowing through R-type channels in hippocampal neurons suggesting that these two channels are complexed together within a calcium nanodomain. 168
Many of the postsynaptic density kinases and phosphatases involved in LTP and LTD, have also been implicated in OD plasticity, including CamKII,169,170 calcineurin, 171 PKA172,173 and Erk kinase, 174 as well as downstream effectors such as CREB. 175 However, their role is still unclear. For instance, it is surprising that CamKII dysfunction, generally thought to affect LTP, affects brief ocular dominance plasticity, a process driven by LTD of deprived eye synapses. 170 While it is possible that this and other discrepancies result from the different function of synapses located in different layers and serving different circuits, it is also possible that the subcellular kinase location and its association with particular calcium signaling domains as well as its spatiotemporal dynamics can affect its contribution to synaptic plasticity. For instance, it has recently been reported that phosphorylation of PSD-95 by CamKII results in PSD reorganization and inhibits spine growth and potentiation of synaptic currents. 176
Another intracellular molecule that is well poised to affect plasticity is actin. The actin cytoskeleton is tethered to the PSD and controls many aspects of PSD organization as well as spine morphology. Signaling within the PSD, on the other hand, is perfectly suited to affect actin polymerization through many different signaling pathways. Actin polymerization is critically involved in spine structural dynamics.93,94,101 It is regulated by many of the same kinases and phosphatases as LTP, LTD and OD plasticity and it's regulation can enhance spine size during LTP and shrink spines during LTD, 177 thereby linking functional and structural aspects of plasticity at the synapse. While intracellular microtubules have traditionally been thought to be compartmentalized to dendrites, recent evidence suggests that dynamic microtubules invade spines and interact with the actin cytoskeleton regulating structural plasticity at dendritic spines. 178 This suggests that the microtubule network may work in concert with actin in the dendritic spine to implement dynamic structural changes during OD plasticity.
Interestingly, the molecular mechanisms that mediate the loss of responses to the deprived eye during MD are not the same as those that mediate recovery from deprivation. While cortical protein synthesis and CREB activation are crucial elements of OD shifts,175,179,180 recovery of visual responses through the re-opened eye does not require either of these mechanisms179,181 and can occur outside the visual critical period. 182 Further, the fact that changes elicited by monocular deprivation can be readily reversed suggests that networks subserving binocular vision are easily reactivated, possibly through the existence of dormant connections or a stable axon scaffold.112,123
Extracellular Signaling
While intracellular pathways of plasticity have been extensively studied, more recently extracellular factors and non-neuronal mechanisms have been implicated in OD plasticity. For instance, signaling by intracortical myelin has been shown to inhibit OD plasticity.
183
Recent studies also suggest that non-traditional cell types, such as astrocytes, are critical for synaptic development, function and plasticity.
184
The importance of astrocytes for visual processing is underscored by the fact that astrocytes show sharply tuned visual responses that are as precisely mapped across the cortical surface as those of neurons and that they can modulate neuronal responses to visual stimuli.
185
Glial-derived tumor necrosis factor-alpha (TNF-
Proteolytic pathways activated by neurons or glia, such as the activation of tissue plasminogen activator (tPA), also appear to be critical for both functional OD plasticity and its associated structural changes at synapses.48,76,187 tPA can be rapidly released from neurons in response to depolarization,188,189 using mechanisms that are also important in LTP-induction 190 and visual plasticity. 191 In fact tPA is up-regulated during LTP, 192 and manipulating tPA activity affects both LTD 193 and LTP. 194 196 Thus tPA can mediate Hebbian processes such as those involved in the loss of deprived eye responses. Additionally, because tPA activity remains high for ∼1 week post-deprivation, 187 it could also mediate slower homeostatic changes. Several downstream effects of tPA activity have been identified as well, though they have yet to be shown to be involved in visual plasticity. tPA plays a role in regulating NMDA receptor kinetics, a known factor in synaptic plasticity. It can cleave the NR1 subunit of the NMDA receptor, increasing calcium influx, 197 and can modulate the NR2B subunit.198,199 tPA can also cleave proBDNF, thus up-regulating BDNF signaling, 200 which affects visual plasticity and synaptic remodeling. 201 Interestingly, tPA can increase extracellular matrix (ECM) proteolysis either directly or through its activation of both plasmin, which is a broad spectrum protease with many possible targets in the ECM, and matrix metalloproteinases (MMPs), 202 which degrade the ECM and play an important role in synaptic plasticity. 203
While the targets of tPA activity in the visual system are not known, several studies suggest that chondroitin sulfate proteoglycans (CSPGs) are cleaved following tPA activation and that these may be candidate extracellular molecules for OD plasticity. For instance, both phosphacan and neurocan can be cleaved by plasmin
Reactivating Plasticity in the Visual Cortex
The molecular mechanisms that control adult OD plasticity are not fully understood, and may be different than those that govern critical period plasticity given the physiological differences 218 and the relative capacities of juvenile and adult cortex for plasticity. 219 However, manipulations of the adult cortex that restore plasticity seem to reinstate known critical period mechanisms of plasticity. Darkrearing adult animals, for instance, reinstates OD plasticity and restores juvenile-like synaptic NMDA and inhibitory signaling profiles in visual cortex. 129 Chemical dissolution of PNNs, which mature during the critical period, also recovers plasticity. 217 Fluoxetine, a selective serotonin reuptake inhibitor, reinstates OD plasticity in the adult rat possibly by decreasing inhibitory drive to developmental levels. 85 While the mechanisms of fluoxetine treatment are not entirely clear, it is interesting to note that fluoxetine has been observed to induce dendritic arborization and spine growth in the hippocampus,220,221 increase synaptic protein expression in a BDNF-dependent manner, 222 enhance cortical axonal remodeling after retinal lesions, 223 affect astrocytic signaling, 224 and change the balance of ECM-remodeling proteinases and their inhibitors. 225 More research is needed in order to elucidate the mechanisms of adult OD plasticity; this will be instructive for understanding the differences between critical period and adult plasticity, as well as for discovering how the transition between the two is determined.
Summary and Future Directions
Competition between the two eyes’ afferents for postsynaptic resources has often been characterized as the primary force behind the OD shift induced by MD. Recent research, however, has described the involvement of a complex framework of spatiotemporal changes at synapses across visual cortex. Some aspects of OD plasticity are directly influenced by the activity or lack thereof in one eye, such as the depression of deprived eye synapses that relies on rapid Hebbian remodeling of the cortical network. Other aspects may be determined by a more prolonged loss of activity and affect synapses globally such as the slower synaptic scaling of responses that may, at least contribute to, the delayed increase in non-deprived eye drive. The implementation of these different mechanisms, however, is diverse and varies across cortical layers and by developmental stage.
Understanding the mechanisms of OD plasticity will require the dissection of physiological, structural and molecular plasticity at individual synapses with identified inputs. One significant advance would be to label each eye's axons separately, allowing their synapses to be differentiated. This might be achieved through transsynaptic viral labeling in eye-specific LGN laminae. From there, each eye's geniculocortical afferents (and the cortical neurons they innervate) would be distinguished by the fluorescent marker they express. 226 Unfortunately this would not provide much information about intracortical circuitry as the diffuse connectivity between cortical neurons makes it more likely that cortico-cortical synapses are binocular. Only the monitoring of synaptic activity through calcium, voltage or molecular activity markers will allow the determination of the binocularity and plasticity at individual intracortical synapses. Existing studies have provided important pieces of this puzzle and allow us to appreciate the complexity of cortical network plasticity. The development of new molecular, optical and electrophysiological tools will facilitate our understanding of how different connections remodel during OD plasticity. As additional work is done, a more detailed picture of local circuit and network level cortical plasticity will certainly continue to emerge.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgements
This work was made possible by grants to A.M. from the Burroughs Welcome Fund, Whitehall Foundation and Sloan Foundation.