
Abstract
Neurodegenerative diseases affect millions of people worldwide, and as the global population ages, there is a critical need to improve our understanding of the molecular and cellular mechanisms that drive neurodegeneration. At the molecular level, neurodegeneration involves the activation of complex signaling pathways that drive the active destruction of neurons and their intracellular components. Here, we use an in vivo motor neuron injury assay to acutely induce neurodegeneration in order to follow the temporal order of events that occur following injury in Drosophila melanogaster. We find that sites of injury can be rapidly identified based on structural defects to the neuronal cytoskeleton that result in disrupted axonal transport. Additionally, the neuromuscular junction accumulates ubiquitinated proteins prior to the neurodegenerative events, occurring at 24 hours post injury. Our data provide insights into the early molecular events that occur during axonal and neuromuscular degeneration in a genetically tractable model organism. Importantly, the mechanisms that mediate neurodegeneration in flies are conserved in humans. Thus, these studies have implications for our understanding of the cellular and molecular events that occur in humans and will facilitate the identification of biomedically relevant targets for future treatments.
Introduction
Neurodegenerative diseases are incurable and severely debilitating conditions resulting from the progressive degeneration of nerve cells. These diseases affect millions of individuals, and as the global population ages, it has been predicted by the World Health Organization that neurodegenerative diseases will overtake cancer to become the second leading cause of death. 1 Extensive research efforts are aimed at tackling the challenges of finding causes, developing cures, and identifying ways to treat those with neurodegenerative diseases. In order to discover cures of neurodegenerative diseases, we must first understand the basic cellular and molecular mechanisms that occur during degeneration of nerve cells.
Neurodegeneration is a tightly controlled and well-orchestrated process resulting in the progressive and nonreversible deterioration of neurons, often culminating in cell death. It requires the function of specific molecular programs that involve cell death machinery and is activated in response to injury, stress, or genetic lesions that induce neurodegenerative diseases. 2 Similar mechanisms also control neurodegeneration during normal synaptic development. An outstanding challenge in the biomedical sciences is to understand the molecular mechanisms that drive neurodegeneration and to discover novel ways to prevent or suppress the progressive nature of neuronal elimination.
In vivo molecular research involving neurodegeneration and neuronal injury inevitably utilizes animal models and historically has focused on vertebrate systems. These models, such as the SOD1 mouse model and chronic sciatic nerve banding, often require complex genetic engineering or surgical manipulations, which have propelled researchers to explore neurodegeneration in more experimentally tractable model organisms, including Drosophila melanogaster. The molecular and cellular hallmarks of neurodegeneration in Drosophila and humans, including a disrupted cytoskeleton, defects in axonal transport, reduced firing capabilities, and subsequent deterioration of the neuron, are similar. 3 Moreover, precise genetic lesions in homologous genes cause comparable disease phenotypes in human beings and flies. Examples include mutations in dynactin, which cause amyotrophic lateral sclerosis in humans and similar severe motor neuron degeneration in Drosophila and mutations in the spectrin/ankyrin skeleton causing motor neuron degeneration in Drosophila and Spinocerebellar ataxia 5 (SCA5) in humans.4–6 The ability to perform large systematic genetic screens in Drosophila has aided in the identification of genes involved in axon and synaptic degeneration.7, 8 Drosophila has additionally facilitated in the identification of genes that suppress the neurodegenerative phenotype, which is of great biomedical relevance. 9
Nerve cells do not undergo cell division and are therefore susceptible to injury via damage by pressure, stretching, or cutting. Neuronal injury can cause defects in axonal transport, synaptic transmission, and/or a complete loss of signal transmission from soma to nerve terminal. When nerve fibers are completely transected, the distal section undergoes sudden and catastrophic disintegration after an initial delay via a process called Wallerian degeneration. Although the timing of axonal fragmentation and disintegration is heterogeneous among the entire axon population, once initiated it is rapid and irreversible. 9 Despite immense efforts directed at understanding the precise molecular pathways giving rise to Wallerian degeneration and ways to suppress it, we still do not know the full molecular mechanisms driving degeneration. Recently, Drosophila has emerged as a good model system to unravel the complex molecular events occurring after neuronal injury.10, 11 However, there is no comprehensive analysis of the temporal order of events that occurs in Drosophila in response to injury that could provide the foundation for future screening/studies.
In this study, we report on an in vivo motor neuron injury assay in Drosophila, which reproducibly induces neurodegeneration by 24 hours post injury. Previously, a similar assay has been utilized to identify specific genes required for neurodegeneration and to dissect transcriptional responses to neuronal injury, but to our knowledge, a foundational temporal observation at the cellular level within axons and at the neuromuscular junction (NMJ) has not been conducted.12–14 In our assay, a simple mechanical injury of segmental nerves causes immediate impairment of the microtubule cytoskeleton and visible disruption of neuroglian, a cell adhesion protein known to play a role in stabilizing neurons. 15 Approximately six hours after injury, there is a buildup of mitochondria indicative of axonal transport defects. Twelve hours post injury, we see an accumulation of ubiquitinated proteins at the NMJ that proceeds synaptic neurodegeneration, which is evident by 24 hours. Our overarching goal is to dissect the spatial and temporal cellular events that occur after neuronal injury, which will lay the framework for future studies in the identification of molecules that prevent or alleviate neurodegeneration.
Materials and Methods
Fly stocks
Drosophila were raised on standard culture media at 25°C. Strains used in these studies include Oregon-R, id118(wild-type); elavclss-GAL416 (neuron-specific); and UAS-mito-GFP (w1118; P{UAS-mito-HA-GFRAP}3, e1 stock #8442 from Bloomington Stock Center).
Neuronal injury and recovery
Second-or early third-instar larvae were injured according to the study by Xiong et al with the following modifications. 12 Larvae were rinsed in cold saline to remove food debris and to decelerate motility. Under a dissecting microscope, larvae were carefully rolled onto their dorsal sides to visualize the segmental nerves through the cuticle prior to pinching ~1/3 of the dorsal cuticle containing the segmental nerves while taking care to avoid the majority of the ventral body wall musculature. After injury, larvae were transferred to a yeasted grape plate covered with a moist Kimwipe and kept alive for specified periods of time at 25°C.
Imaging and analysis
Images were digitally captured using EZ-C1 Nikon software on a Nikon D-Eclipse CI laser scanning confocal microscope and analyzed using Fiji software. Individual nerves and synapses were optically sectioned at 0.5 µm with a Plan Apo 100×/1.40 oil immersion objective. Using Fiji software, Z-stacks were combined into a single maximum projection image. Accumulations of ubiquitinated proteins were analyzed in Fiji using the Particle Analyzer function, set to pick up puncta >10 pixels in size. Two-tailed unpaired t-tests and nonparametric two-tailed Wilcoxon-Mann-Whitney tests were performed with 95% confidence intervals using GraphPad Software.
Immunohistochemistry
At 0, 6, 12, 24, and 48 hours after mechanical neuronal injury, larvae were dissected in phosphate-buffered saline and fixed in either 4% paraformaldehyde in PBS for 20 minutes to visualize green fluorescent protein or in Bouin's fixative for two minutes for all other antibodies and stained according to standard procedures.8, 17 Primary antibodies were used at the following dilutions: 1:100 anti-Bruchpilot (nc82; Developmental Studies Hybridoma Bank), 1:100 anti-BP104 (neuroglian; Developmental Studies Hybridoma Bank), 1:20 anti-Futsch (22C10; Developmental Studies Hybridoma Bank), 1:10,000 anti-Dig 18 (anti-discs large), and 1:500 anti-FK2 (Millipore). Secondary Alexa Fluor antibodies (goat anti-mouse 488, goat anti-rabbit 555, and goat anti-rabbit 647) were obtained from Jackson ImmunoResearch Laboratories, Inc. Cy3-and Cy5-conjugated horseradish peroxidase (HRP) were also obtained from Jackson ImmunoResearch Laboratories, Inc. and were used at a 1:300 dilution.
NMJ degeneration quantification
The neurodegenerative phenotype was analyzed according to standard protocols.8, 17, 19–23 Neurodegeneration at the NMJ was scored at 400× magnification with the observer being blind to the genotype and injury conditions. Muscle 6/7 of segments A2-A6 were quantified with a degenerative event being defined as an area of the NMJ containing clearly defined postsynaptic Dig staining without any apposing presynaptic Brp staining. Two-tailed unpaired t-tests and nonparametric two-tailed Wilcoxon-Mann-Whitney tests were performed with 95% confidence intervals using GraphPad Software.
Larval motility movies
Drosophila third-instar larval motility was recorded in 30-second intervals using Moticam 2.0 software connected to a Moticam 3.0 MP digital camera mounted on a dissecting microscope set at 40× magnification. Using QuickTime player software, the video clips were cropped to demonstrate the crawling behavior of a single larva across one field of view.
Results
Mechanical injury causes immediate disruption of the microtubule cytoskeleton and the L1-type cell adhesion molecule neuroglian resulting in disrupted axonal transport
Neuronal injury was induced by mechanically damaging peripheral nerves in Drosophila larvae with size 5 forceps. To ensure that each animal experienced comparable mechanical injury, we first examined the characteristic crawling behavior of each larva. Uninjured larvae move by sequentially executing a single mouth hook extension followed by an initiation of a peristalsis-like body wall contraction, which drives the animal forward (Supplementary Movie 1). Immediately after mechanical injury, however, larvae showed distinctive paralysis of all body wall musculature posterior to the site of injury and were thus unable to initiate posterior body wall contractions (Supplementary Movie 2). Despite the severe paralysis, injured larvae went through normal pupation and were able to survive into adulthood.
In Drosophila, antibodies against HRP act as specific neuronal membrane markers allowing for visualization of all neurons, including peripheral motor neuron axons.
24
In uninjured third instar larvae, fluorescently conjugated HRP antibodies were employed to visualize the series of paired segmental nerves exiting both sides of the ventral nerve cord and extending along the length of the animal. Our neuronal injury assay resulted in damage to ~6-12 segmental nerves depending on the precise location of the crush site, as indicated by a reduction in the amount of HRP staining immediately after injury (Fig. 1, boxed area). Sites of injury range from ~100 to 200 µm in length.
Mechanical Injury of Drosophila Larvae Demonstrates Damage to the Majority of Segmental peripheral Nerves. (
Each segmental nerve in Drosophila third instar larvae contains between 60 and 80 individual axons that extend from their cell bodies, which reside in the ventral nerve cord.
25
Motor neuron axons within segmental nerves can be visualized by staining for neuronal membranes using HRP and for axonal microtubules using antibodies against the Drosophila specific gene, Futsch (MAP1B). Futsch is expressed only in neuronal cells, has sequence similarity to vertebrate neurofilament proteins, and has been used extensively to visualize neuronal projections in Drosophila.
26
Uninjured animals show numerous continuous axonal microtubules with intact neuronal membranes within each segmental nerve extending from the dorsal nerve cord. However, immediately after injury, both ends of each neuronal crush site contain severely disrupted axonal microtubules. Although some individual axonal microtubules extend farther into the crush site, the majority of microtubules become completely disintegrated as they extend into the epicenter of the neuronal crush site (Fig. 2).
The Microtubule Cytoskeleton is Severely Disrupted Upon Mechanical Injury to Motor Neurons. (
In addition to severe disruption of the axonal microtubule cytoskeleton, neuroglian, an L1-type cell adhesion molecule (L1-CAM), is altered immediately after mechanical injury to segmental nerves.27, 28 Drosophila neuroglian was first discovered in 1989 as an essential integral membrane protein localizing to neuronal cell bodies and all along neuronal axons and more recently has been shown to be essential for synaptic stabilization.28, 29 In wild-type uninjured segmental nerves, neuroglian extends the length of each individual axon as visualized by colocalization with the neuronal membrane marker, HRP. However, after neuronal injury, there is a complete breakdown of the organized axonal neuroglian near the ends of each crush site, resulting in a complete obliteration of neuroglian within the actual injury site (Fig. 3).
Injured Segmental Nerves Display Disordered Neuroglian Organization at Sites of Injury. (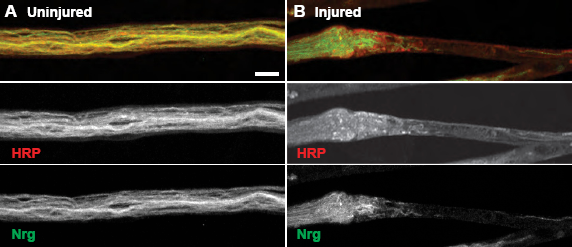
Severe disruption of microtubules within motor neuron axons prevents typical anterograde and retrograde axonal transportation. Interestingly, axonal transport defects are observed in, and thought to contribute to, the pathologies of neuronal injury and a diverse range of neurodegenerative diseases.
30
Drosophila have long served as an excellent model system to study the mechanisms of axonal transport due to the presence of larval segmental nerves containing individual axons extending from the central nervous system down the length of the animal to their synaptic terminals along the body wall musculature.25, 31 To examine the effects of mechanical injury on axonal transport, we focused on mitochondria, which are transported along larval motor axons in the anterograde direction by kinesin-1 and to the synapse in the retrograde direction by cytoplasmic dynein where they regulate synaptic strength.32, 33 Uninjured segmental nerves contain a relatively uniform number of mitochondria dispersed along motor neuron axons as demonstrated by examining mitochondria tagged with green fluorescent protein (mito-GFP). However, after injury of segmental nerves, there is a buildup of mito-GFP at the proximal and distal ends of the crush site within six hours indicating that axonal transport is inhibited. Additionally, there appears to be no mito-GFP within the crush site itself at any point after the initial injury (Fig. 4). These results are consistent with a previous study in Drosophila demonstrating that neuronal injury causes vesicular cargo accumulations at sites of axonal injury with an absence of cargo within the crush site itself.
12
Mechanical Injury of Segmental Nerves Disrupts Axonal Transport and Leads to a Buildup Of Mitochondria at the Proximal End of the Crush Site. (
Ubiquitinated proteins accumulate at the NMJ ~12 hours after mechanical injury prior to neurodegeneration
Ubiquitinated protein accumulations have long been known to be associated with neurodegenerative diseases.
34
We sought to examine if neuronal injury of Drosophila motor neurons could induce increases in the amount of ubiquitinated proteins at the NMJ using an antibody (α-FK2) against both mono-and poly-ubiquitinated proteins. Quantification of numbers of FK2-positive puncta within the NMJ at muscle 6/7 revealed a significant increase 12 hours post injury. Uninjured larval NMJs contained an average of 0.19 puncta per synapse (n = 13 NMJs), whereas injured larvae had an average of 6.55 puncta per synapse (n = 20 NMJs; Fig. 5; P < 0.0001). These data are consistent with previous reports of autopsies from various human spinal cord traumas signifying that ubiquitin accumulations mark an early event in neuronal injury.
33
Injury to Motor Neuron Axons Induces a Significant Increase in Ubiquitinated Proteins at the NMJ. (
It has been previously reported in Drosophila that segmental nerve injury causes a significant loss of presynaptic vesicular glutamate transporter (VGlut) proteins at NMJ synapses.
13
We wanted to examine if other presynaptic markers were lost after injury and to examine the extent of neurodegeneration using our previously established method for quantitatively investigating neurodegeneration at the Drosophila NMJ.2, 17, 19–23 In this assay, NMJs were stained for the presynaptic active zone marker Bruchpilot (Brp; stained with nc82 antibody) and the postsynaptic marker discs large (Dig) at various time points after mechanical injury. Boutons clearly stained with Dig but lacking nc82 immunoreactivity signify a neurodegenerative event. The NMJs from uninjured animals show the presynaptic marker Brp in perfect apposition with the postsynaptic marker Dig (Fig. 6A). However, 24 hours post injury, NMJs exhibited various degrees of missing presynaptic Brp staining at the NMJ ranging from moderate neurodegeneration (10 boutons retracted) to severe neurodegeneration (>10 boutons retracted; Fig. 6B and C). The highly compact muscle membrane folds that create the postsynaptic part of the NMJ degenerate more slowly then their presynaptic counterpart and thus persist beyond the neurodegenerative events that we are characterizing.
19
The severity of neurodegeneration was quantified as the average number of boutons per NMJ exhibiting degeneration and was significantly higher in animals with neuronal injury (average = 4.44, n = 237 NMJs) compared to uninjured animals (average = 0.07, n = 140 NMJs; Fig. 6D; t-test: P = 0.030; significance remains with Wilcoxon-Mann-Whitney test: P = 0.0007). The frequency of neurodegeneration was quantified as the average percentage of NMJs per animal with degeneration and was determined to be significantly higher after neuronal injury in animals (average = 10.33, n = 25 animals) compared to in uninjured animals (average = 0.14, n = 14 animals; Fig. 6E; Mest: P = 0.0018; significance remains with Wilcoxon-Mann-Whitney test: P = 0.0016). To more fully understand the neurodegenerative frequency, we also measured the percentage of NMJs with degeneration of >1 bouton retracted (average = 11.20) and 3 boutons retracted (average = 4.49; Fig. 6F; t-test: P = 0.0286; significance remains with Wilcoxon-Mann-Whitney test: P = 0.0429). This neurodegenerative phenotype is less severe than what is seen by genetic modifications but is similar to previously published results examining the loss of presynaptic VGlut proteins at the NMJ after neuronal injury.2, 13, 17, 19–23 Together, these data suggest a spatial and temporal sequence of cellular events originating at the site of axonal injury with immediate cytoskeletal defects inducing axonal transport dysfunction by six hours, followed by accumulations of ubiquitinated proteins by 12 hours and subsequent neurodegeneration at the NMJ by 24 hours (Supplementary Table 1).
Neuronal Injury Induces Moderate-to-Severe Neurodegeneration at the NMJ of Muscle 6/7. (
Discussion
In humans, injury of peripheral motor neurons results in reduced sensation, reduction of innervation density, and often a very poor prognosis for functional outcomes. In order to develop novel therapeutic interventions following neuronal injury, we must first gain a better understanding of the cellular and molecular mechanisms leading to the observed neurodegenerative phenotypes. This work provides a spatial and temporal framework for the cellular events following neuronal injury in a simple reproducible Drosophila model system. We have demonstrated that after mechanical injury there is an immediate disruption of both microtubules and neuroglian followed by a buildup of mitochondria at six hours signifying axonal transport defects. Accumulations of ubiquitinated proteins at the NMJ occur by 12 hours, which proceeds neurodegeneration observed by 24 hours (Supplementary Table 1). Providing this initial groundwork will allow for a more comprehensive dissection of the molecular mechanisms triggering these processes and for the exploration of novel means to suppress the devastating outcomes resulting from neuronal injury.
The peripheral nervous system contains axons that extend great distances from their cell bodies and are therefore susceptible to damage in multiple cellular compartments. In most cases, injury of neurons occurs within axons but activates molecular signaling pathways at a distance (i.e. within the cell body or at the synaptic terminal). In humans, axonal injury is universally found in spinal and head trauma and has been recognized as a key predictor of patient prognosis. 35 One of the major consequences of axonal injury is disruption of axonal transport, which contributes to the pathologies of many neurodegenerative diseases. 30 In peripheral nerves, an intact axonal microtubule cytoskeleton is crucial for normal axonal transportation and synaptic transmission at downstream NMJs. In our model, mechanical injury induces an immediate disruption of the microtubule cytoskeleton extending the distance of the actual crush site itself. We observe, as others have, that a primary effect of axonal injury is the deformation of axonal microtubules resulting in the interruption of axonal transport and the buildup of transported materials within hours of trauma. 36
In addition to immediate disturbance of the microtubule cytoskeleton, we find that neuroglian is also significantly inhibited at neuronal injury sites. Neuroglian, an L1-CAM, is required for the stabilization of basic neuronal cytoskeletal architecture and for maintenance of the overall stability of neurons and dendrites in Drosophila.15, 28, 37, 38 Mutations in the human L1-CAM gene are also responsible for a range of neurodegenerative disorders, including spastic paraplegia type I.39, 40 In Drosophila, neuroglian acts as key regulator of synapse stability in both the central and peripheral nervous systems by linking neuronal membrane proteins to the underlying spectrin skeleton. 28 Interestingly, spectrin has previously been demonstrated to be essential for synaptic stability and lack of spectrin results in severe neurodegeneration at NMJs.21, 41 It is therefore formally plausible that mechanical injury and subsequent disruption of neuroglian may result in neurodegeneration simply due to the disruption of spectrin at the injury site.
Accumulations of ubiquitinated proteins are known to be a common characteristic of many human neurodegenerative diseases; however, the mechanistic role remains elusive.42, 43 It remains unclear if these protein aggregates are toxic or rather are a byproduct of neurons sequestering toxic or damaged proteins. 44 In healthy neurons, rapid degradation by the ubiquitin-proteasome system (UPS) prevents high levels of ubiquitinated proteins from accumulating and it has been suggested that neuronal injury and neurodegenerative diseases have reduced UPS activity due to the presence of toxic microenvironments and/or mitochondrial dysfunction. 45
Although aggregations of ubiquitinated proteins proceed neurodegeneration in certain genetic backgrounds, the delayed accumulation of ubiquitinated proteins at a distance from the injury site prior to synaptic degeneration is interesting and to our knowledge has not been previously described.46, 47 It is known that inhibition of the UPS prevents developmental axon pruning and delays injury-induced axon degeneration, possibly through stabilization of disrupted microtubules.48, 49 The UPS, and thus protein turnover, is known to be under precise control at synapses to regulate processes ranging from synaptogenesis to plasticity and remodeling. 50 Our simple genetic model for neuronal injury may provide a framework from which to determine: 1) how and why ubiquitinated proteins accumulate at the NMJ after axonal injury, 2) if axonal injury disrupts the UPS or if it is functioning properly but overloaded due to the shear number of damaged proteins, and 3) whether ubiquitinated protein accumulations at the NMJ can serve as a biomarker for neurodegeneration.
This research lays the groundwork for future studies involving the molecular and cellular basis of neurodegeneration after motor neuron injury. Despite the apparent time-scale differences between slowly progressing human neurodegeneration and our acute neuronal injury model, we believe that the model presented here may produce comparable pathological changes as that of humans. Subsequent studies using this simple, genetically tractable model system to study axonal injury will hopefully shed light on numerous aspects that are still unclear such as the involvement and activation of glia, the role of calcium and mitochondria, and the role neurodegeneration plays in muscle atrophy. Recent technological advances in microfluidics may aid in answering these and other remaining questions related to axonal injury and the subsequent degeneration at the NMJ. 14
Conclusion
Together our findings and those of others identify Drosophila as an excellent genetically tractable model system to study motor neuron injury and neurodegeneration. The ability to temporally examine the cellular deterioration of neurons after injury provides insight into the molecular mechanisms that drive the degenerative process. In particular, our study highlights the accumulation of ubiquitinated proteins prior to degeneration, which may offer an early indication of neurons that are in peril. We hope that gaining an understanding of how neurons degenerate may also assist in the discovery of novel ways to suppress the active degenerative signaling process. There is precedence for suppression of degeneration in Drosophila motor neurons, which someday may extend to novel therapeutic discoveries in humans. 8
Author Contributions
Conceived and designed the experiments: BLL and LCK. Analyzed the data: BLL, SHA, and LCK. Wrote the first draft of the manuscript: LCK. Contributed to the writing of the manuscript: LCK. Agreed with manuscript results and conclusions: BLL, SHA, NF, RF, and LCK. Jointly developed the structure and arguments for the paper: BLL, SHA, NF, RF, and LCK. Made critical revisions and approved the final version: BLL, SHA, NF, RF, and LCK. All the authors reviewed and approved the final manuscript.
Supplementary Materials
Supplementary movie 1
A third instar wild-type larva has a characteristic crawling motility in which a single mouth hook extension is followed by a body wall contraction propelling the larva forward.
Supplementary movie 2
A third instar wild-type larva shortly after being subjected to neuronal injury. The characteristic crawling motility is severely disrupted with the posterior third of the larva unable to perform body wall contractions.
Supplementary table 1
Temporal sequence of cellular events leading to neurodegeneration after mechanical injury at the NMJ.
Footnotes
Acknowledgments
We would like to thank all members of the Keller and Magie Labs at Quinnipiac University for helpful experimental suggestions and for providing a stimulating and interactive laboratory environment for undergraduate research. For fly lines, we thank the Bloomington Stock Center (Indiana University), and for antibodies, we thank the Developmental Studies Hybridoma Bank (University of Iowa).