
Abstract
Neutrophils (also called polymorphonuclear leukocytes, PMNs) are the most abundant white blood cells in humans and play a central role in innate host defense. Another distinguishing feature of PMNs is their short lifespan. Specifically, these cells survive for less than 24 hours in the bloodstream and are inherently pre-programed to die by constitutive apoptosis. Recent data indicate that this process is regulated by intracellular signaling and changes in gene expression that define an “apoptosis differentiation program.” Infection typically accelerates neutrophil turnover, and as such, phagocytosis induced cell death (PICD) and subsequent clearance of the corpses by macrophages are essential for control of infection and resolution of the inflammatory response. Herein we reprise recent advances in our understanding of the molecular mechanisms of neutrophil apoptosis with a focus on regulatory factors and pathway intermediates that are specific to this cell type. In addition, we summarize mechanisms whereby perturbation of PMN death contributes directly to the pathogenesis of many infectious and inflammatory disease states.
Keywords
Introduction
Polymorphonuclear leukocytes (PMNs) are the most abundant leukocyte in human blood, 1 survive for less than 24 hours in circulation, and are generated in the bone marrow at a rate of 1011 cells/day. 2 This large-scale production must be matched by equivalent removal to maintain homeostasis. To achieve this, circulating neutrophils die via constitutive apoptosis, and are cleared by macrophages in the liver, spleen, and bone marrow. 3 PMN abundance is in keeping with the fact that these cells provide a critical first line of defense against infection during the innate immune response. Typically, neutrophils are rapidly recruited to sites of infection and inflammation where they ingest invading microbes and deploy NADPH oxidase-derived reactive oxygen species (ROS), proteases, and antimicrobial peptides that act in concert to create a highly lethal intraphagosomal environment. 4
Failure to properly regulate neutrophil abundance and turnover directly causes or contributes to human disease. Neutrophilia is the accumulation of neutrophils and their histotoxic cargo in the tissues, which has been associated clinically with dyspnea, hypoxia, and neurologic deficits, possibly because of increased blood viscosity and obstructed blood flow.5,6 Conversely, neutropenia markedly impairs innate defense and increases susceptibility to infection.4,7 Precise regulation of PMN apoptosis is also essential for resolution of inflammation as this prevents release of toxic intracellular components that can damage healthy tissue. 8 In addition, efferocytosis of dying PMNs dampens proinflammatory cytokine production and reprograms macrophages to a pro-resolution phenotype that favors restoration of tissue homeostasis.9–11 In keeping with this, defects in PMN turnover and clearance are indicative of a dysregulated and defective inflammatory response that can enhance tissue destruction. 8 Herein, we review the molecular mechanisms of neutrophil apoptosis with a focus on recent discoveries and pathways specific to this cell type. The ability of pro-survival signaling to transiently delay apoptosis during neutrophil extravasation is also discussed. Finally, we summarize our current understanding of the mechanisms by which aberrant PMN turnover contributes to infectious and inflammatory disease.
Conserved Apoptosis Machinery and Pathways
Apoptosis is a conserved mechanism of programed cell death. Two main apoptotic pathways have been described and are present in nearly all cell types, including PMNs. The extrinsic pathway is activated by ligation of surface death receptors that bind Fas ligand, TNFα, or TRAIL. In contrast, the intrinsic pathway is regulated at the level of mitochondria and is initiated by disruption of the outer mitochondrial membrane, which releases cytochrome C and other proapoptotic factors into the cytosol (Fig. 1). A third apoptotic pathway, phagocytosis-induced cell death (PICD), couples microbe killing to accelerated neutrophil death at sites of infection.12–14 Regardless of the specific pathway activated, apoptotic PMNs undergo characteristic morphological changes that include membrane blebbing, cell body shrinkage, cytoplasmic vacuolation, and nuclear condensation. 15 DNA fragmentation and loss of plasma membrane asymmetry, with accumulation of phosphatidylserine (PS) in the extracellular leaflet, are additional endpoints that can be used to quantify apoptosis kinetics, and the surface-exposed PS facilitates recognition of dying PMNs by macrophages and their internalization via a process called efferocytosis. 16 Although our understanding of the molecular regulation of apoptosis is rapidly evolving and is noteworthy for its increasing complexity, whether a neutrophil lives or dies is governed at its core by the relative abundance of pro-survival and proapoptotic regulatory factors. In aging PMNs, expression of pro-survival factors wanes and pro-death factors accumulates, and this imbalance is critical for normal turnover via constitutive apoptosis.
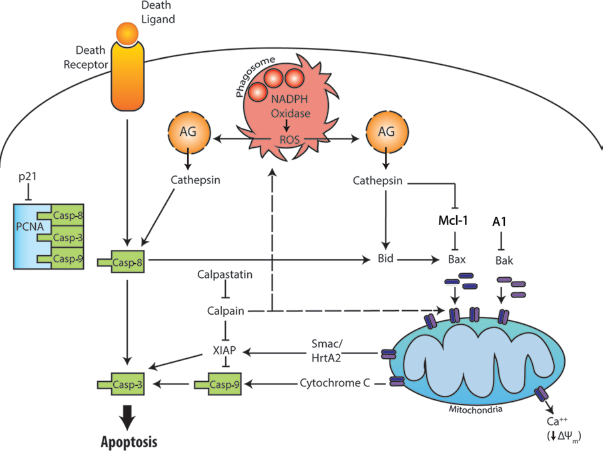
Overview of the extrinsic, intrinsic, and phagocytosis-induced apoptotic pathways. The extrinsic pathway is initiated by ligation and clustering of plasma membrane death receptors leading to activation of caspase-8 and caspase-3. The intrinsic apoptotic pathway is initiated when the relative abundance of the proapoptotic Bcl-2 family members (Bax and Bak) exceeds that of their anti-apoptotic counterparts (Mcl-1 and A1), which allows Bax and Bak to oligomerize and form pores in the outer mitochondrial membrane. Release of cytochrome C into the cytosol is an essential signal for activation of caspase-9 upstream of caspase-3. The PICD pathway is driven by particle uptake and NADPH oxidase-derived ROS. Leakage of cathepsins from damaged azurophilic granules leads to death receptor-independent activation of caspase-8. In addition, cathepsin-driven activation of Bid and degradation of Mcl-1 favor mitochondrial disruption and intrinsic pathway activation. Additional details are provided in the text.
Caspases
Caspases are a family of cysteine proteases with roles in apoptosis, inflammation, and development. Caspase-8 and caspase-9 are the initiator caspases of the extrinsic and intrinsic apoptosis pathways, respectively, that in turn mediate activation of the executioner caspase-3 to evoke many of the defining biochemical and biophysical changes that occur during apoptosis. 15 In healthy cells, caspases reside in the cytosol as inactive pro-enzymes and, as described below, their inactive state is maintained by direct association with molecules of the inhibitor of apoptosis protein (IAP) family.
The extrinsic pathway
Ligation and oligomerization of Fas, TNF receptor 1, or the TRAIL receptor triggers formation of a receptor-associated death-inducing complex for recruitment and activation of caspase-8. Although it is clear that death receptor ligands can trigger PMN death, the extrinsic pathway in this cell type is noteworthy for several reasons. First, Scaffidi et al categorized cells into two distinct groups based on their respective responses to Fas ligand. 17 In this regard, Type I cells exhibit rapid caspase activation that is independent of mito- chondrial permeabilization. However, in Type II cells, including PMNs, the extrinsic pathway is not sufficient for cell death, and apoptosis requires signal amplification via the intrinsic pathway. These two pathways are linked by Bid, which is truncated by caspase-8 and then is translocated to mitochondria. In this locale, truncated Bid facilitates oligomerization of Bax and Bak, two proteins that directly disrupt the outer mitochondrial membrane, a critical control point for initiation of the intrinsic apoptosis pathway. 18 Second, the effects of TNFα on neutrophil viability are dose dependent such that low levels of this cytokine (0.1 ng/mL) prolong cell lifespan, whereas high doses (10 ng/mL or more) trigger apoptotic death. 19 NADPH oxidase-derived ROS are also essential for TNFα-mediated apoptosis, and as such this pathway is absent in persons with chronic granulomatous disease (CGD). 19 Finally, the extrinsic pathway appears to play little or no role in constitutive PMN turnover despite clear evidence of caspase-8 processing and activation under these conditions, and the ability of PMNs to synthesize and secrete Fas ligand.17,20–22 Indeed, the data suggest that caspase-8 activation in aged neutrophils occurs by a mechanism that is independent of death receptor ligands. 21 Consistent with this, inhibition or deletion of Fas does not alter constitutive PMN turnover. 23 Nonetheless, the extrinsic pathway, including Fas, plays a more important role in neutrophil apoptosis during infection and inflammation.24,25
The intrinsic pathway
The intrinsic pathway is critical for both constitutive and stimulated PMN apoptosis. Although the intracellular signal that initiates death of aging neutrophils remains obscure, a key early event in this process is mitochondrial outer membrane permeabilization (MOMP).25–27 Of note, PMNs contain relatively few mitochondria and rely on glycolysis for ATP generation, and for this reason, it has been suggested that the role of mitochondria in neutrophils is restricted to apoptosis. 28 At the molecular level, MOMP is mediated by two pro-death members of the Bcl-2 family, Bax and Bak. As shown in Figure 1, these proteins oligomerize and are inserted into the outer mitochondrial membrane, which disrupts organelle membrane potential and also mediates rapid release of cytochrome C, Smac/DIABLO, and HtrA2/Omi from the intermembrane space into the cytosol. Together with apoptosis protease activating factor (APAF), cytochrome C forms a complex called the apoptosome that recruits and activates procaspase-9. At the same time, Smac/ DIABLO and Htra2/Omi indirectly favor activation of intrinsic pathway caspases by binding and inactivating various pro-survival factors. 27 These and other key regulatory factors are discussed in the next section.
Bcl-2 family proteins and IAPs
MOMP is tightly controlled by the ratio of pro-survival and pro-apoptosis proteins of the Bcl-2 family. 29 Neutrophils contain only two pro- survival Bcl-2 proteins, Mcl-1 and A1 (also called BCL2A1 or Bfl-1), of which Mcl-1 in particular is essential for cell viability.30,31 In contrast, immature neutrophils, HL-60 cells, and most other cell types also contain Bcl-2 and Bcl-XL, but these factors are downregulated at the mRNA and protein levels during neutrophil differentiation.32–34 Mcl-1 and A1 are short-lived and must be continually synthesized to prevent cell death. 34 Consistent with this, Mcl-1 levels decline with PMN age and correlate closely with the kinetics of apoptosis. 35 Conversely, proapoptotic Bcl-2 family proteins (Bax, Bak, and Bid) have long half-lives. In healthy PMNs, Mcl-1 and A1 are present in excess and prevent mitochondrial disruption by Bax, Bak, and Bid via various mechanisms that include direct contact-mediated inhibition and regulation of phosphorylation state.29,31
Another important group of regulatory factors is the IAPs (XIAP, cIAP1, cIAP2) that impair caspase activity. 36 In particular, XIAP binds directly to and inhibits procaspase-9 and -3 processing and activity, and may be the most important IAP in neutrophils.36,37 During apoptosis, XIAP is cleaved and inactivated by Smac/DIABLO and HtrA2/Omi after their release into the cytosol, or by the cysteine protease calpain.38–40 As calpain is constitutively active, cleavage of calpain substrates is prevented in healthy cells by its endogenous inhibitor calpastatin. 37 In contrast, cIAP1 and cIAP2 affect caspase activity indirectly via association with TNF receptor complexes and protein ubiquitination.41,42
Neutrophil-Specific Regulatory Factors
Cyclin-dependent kinases(CDKs)
Expression of most CDK isoforms is downregulated during neutrophil differentiation. Until recently, residual expression of CDK7 and CDK9 in mature neutrophils was considered irrelevant, consistent with the fact that these cells are post-mitotic. 43 However, it is now appreciated that these enzymes are important for PMN viability as the CDK inhibitors R-roscovitine and 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole can induce neutrophil apoptosis and reduce neutrophil numbers in animal models of inflammation.43–45 Additional studies are needed to define CDK substrates and to define precisely their role in modulation of PMN lifespan, but preliminary studies suggest that RNA polymerase II may be a direct target. 45
Proliferating cell nuclear antigen (PCNA)
In most cell types, PCNA is a nuclear protein that participates in DNA synthesis and repair. In marked contrast, PCNA is found exclusively in the cytoplasm of neutrophils, and in this locale functions as a pro-survival mediator that binds constitutively to procaspases-3, -8, and -9 and blocks their proteolytic processing and activation. 46 Accordingly, PCNA levels decline during neutrophil apoptosis and are sustained by pro-survival stimuli, such as G-CSF. 46 Furthermore, exogenously induced changes in PCNA levels can alter neutrophil susceptibility to both intrinsic and extrinsic apoptotic pathway stimuli. 46 Taken together, these data suggest that PCNA is a key player required for neutrophil survival. 47 Binding partners of PCNA in other cell types include p21, a nuclear protein and endogenous CDK inhibitor that impairs DNA replication. 47 Nonetheless, p21 levels are low in PMNs and whether PCNA-p21 interactions modulate neutrophil survival is as yet unknown. 47
Myeloid nuclear differentiation antigen (MNDA)
MNDA is a recently identified regulatory factor that relocalizes from the nucleus to the cytosol in aging PMNs, is cleaved by caspases, and directly associates with Mcl-1 to promote its degradation via the proteasome.48,49 In contrast, MNDA is retained in the nucleus in cells exhibiting prolonged viability, such as during sepsis or following treatment with LPS or platelet-activating factor.48,49
The Neutrophil Apoptosis Differentiation Program and PICD
Our understanding of the role of gene expression in neutrophil turnover was revolutionized by DeLeo and colleagues, who were the first to demonstrate in an elegant series of studies that constitutive neutrophil apoptosis requires changes in gene expression that collectively comprise an apoptosis differentiation program.12,13,50,51 Before this work, it was generally believed that mature PMNs were transcriptionally inert. However, it is now clear that apoptosis is the final stage of the neutrophil lifecycle that is controlled by gene expression as well as intracellular signaling. In addition, these studies provided a mechanism to account for the ability of phagocytosis to profoundly accelerate neutrophil death, an observation that was made first by Watson et al in 1996. 52 The PICD pathway is triggered by phagocytosis and NADPH oxidase-derived ROS, and also requires global changes in gene expression.13,53–55
Although the individual genes of interest are too numerous to list here, pathway analysis indicates that metabolism, cytokine production, receptors, signal transduction, transcription, and host defense are affected, in addition to apoptosis and cell survival. Overall, there is a general downregulation of proinflammatory capacity, and nearly all critical PMN functions are markedly impaired including chemotaxis, phagocytosis, oxidant production, and degranulation.12,13,50,51,56–58
Of note, PICD is triggered following phagocytosis of complement- or antibody-coated particles as well as bacteria, and has been documented in PMNs infected with Escherichia coli, Listeria monocytogenes, Burkholderia cepacia, Streptococcus pyogenes, and Borrelia hermsii.13,53 However, PICD does not occur in neutrophils from persons with CGD because of the lack of NADPH oxidase-derived ROS, 54 and under these conditions, PMN proinflammatory capacity is sustained, and host tissue damage and destruction are enhanced.8,14,54
Effects of Pro-Survival Signaling on Neutrophil Apoptosis and Lifespan
Several partially overlapping signaling pathways associated with activation of phosphatidylinositol 3-kinase (PI3K) and Akt, ERK, and NFkB act downstream of growth factor receptors, adhesion receptors, and TLR4 to temporarily delay apoptosis as a means to ensure the viability of neutrophils as they migrate from the bloodstream into infected and inflamed tissues.14,59,60 In this regard, the effects of G-CSF, GM-CSF, and LPS have been studied extensively.14,59,61 PI3K/Akt signaling is common to all these pathways and plays a pivotal role in neutrophil survival. 62 Akt is a serine/threonine kinase that is activated by the lipid products of class I PI3Ks. Directly relevant to apoptosis, Akt-mediated phosphorylation inactivates caspase-9 and prevents Bax association with mitochondria. 62 Inactivation of another Akt target, GSK3β, prevents phos- phorylation, ubiquitination, and proteasomal degradation of Mcl-1. 62 The MAP kinase ERK is activated in PMNs treated with GM-CSF, 63 G-CSF, 64 or LPS, 65 and increases Mcl-1 66 and XIAP 67 protein stability to prolong neutrophil lifespan. Sites of infection and inflammation are often characterized by low oxygen tension, and the fact that hypoxia prolongs PMN survival in conjunction with ERK activation underscores the ability of the local tissue microenvironment to modulate PMN viability and function.59,68 In contrast, the effects of p38 MAP kinase on PMN survival and apoptosis are uncertain because of conflicting data, and may be context dependent.67,69–71 Pro-survival signaling also leads to NF-kB activation, and downstream targets of this pathway directly relevant to PMN apoptosis include the genes that encode A1, A20, XIAP, cIAP1, cIAP2, and Mcl-1, and the anti-oxidant MnSOD.20,72
Manipulation of PMN Apoptosis by Microbial Pathogens
In the past few years, our understanding of the ability of bacterial, fungal, parasitic, and viral pathogens to manipulate PMN turnover has increased dramatically. In general, these effects fall into at least three categories. Some pathogens delay PMN death to maintain viability of their replicative niche, whereas others accelerate apoptosis or trigger PMN lysis to evade intracellular killing. Another variation on this theme is the use of infected neutrophils as Trojan horses for infection of macrophages. Recent advances in this exciting field are summarized in Figure 2 and discussed below.
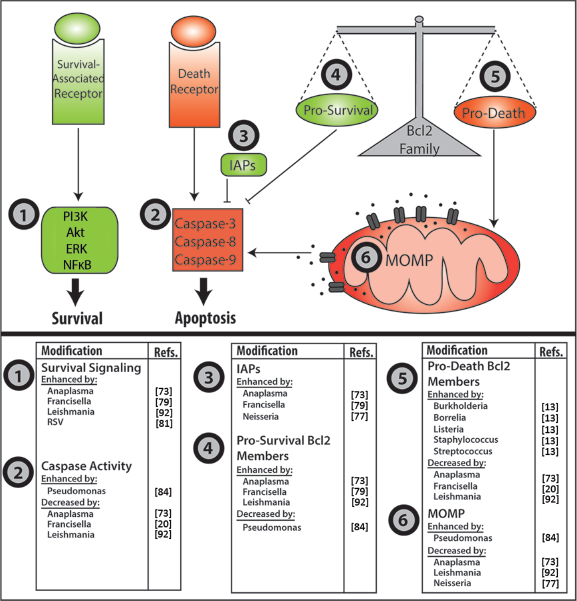
Pathogen manipulation of neutrophil apoptosis. Major control points of neutrophil apoptosis that are targeted by microbial pathogens are depicted in the upper panel. Organisms known to affect these steps are listed in the lower panels. MOMP, mitochondrial outer membrane permeabilty.
Extending neutrophil lifespan for intracellular growth
Anaplasma phagocytophilum is an obligate intracellular pathogen of neutrophils, and a large body of data indicates that this organism uses a multifaceted strategy to modulate multiple apoptotic and survival signaling pathways in PMNs. 73 Coincident with large-scale changes in gene expression of infected cells, XIAP and A1 levels increase, whereas expression of death-promoting factors (such as Bid and Bax) declines.73,74 As depicted in Figure 1, these changes preserve mitochondrial integrity, and as such diminish and delay caspase-3 activation. A. phagocytophilum also stimulates pro-survival signaling via p38 MAPK, Akt, ERK, and NFkB. 73 At the same time, blockade of NADPH oxidase activation and downregulation of Bax likely synergize to impair activation of PICD.73,74 Although the bacterial factors that direct these events are incompletely defined, a role for the type IV secretion system is established, whereby the secreted effector protein Ats-1 translocates to mitochondria and sustains organelle integrity. 73
Another facultative intracellular bacterium, Chlamydia pneumoniae, delays apoptosis by stimulating neutrophil secretion of IL-8, 75 as does the fungal pathogen Paracocciodes brasiliensis. 76 Neisseria gonorrhoeae transiently delays the onset of apoptosis in parallel with upregulation of cIAP2 and XIAP, despite stimulation of NADPH oxidase activation during phagocytosis of this important human pathogen. 77
Recently, we demonstrated that the facultative intracellular bacterium responsible for the disease tularemia, Francisella tularensis, markedly delays neutrophil apoptosis; 20 and we hypothesize that this defect in cell turnover contributes to the robust neutrophil accumulation, granuloma formation, and necrotic tissue damage that are characteristic of this disease. F. tularensis uses multiple mechanisms to inhibit NADPH oxidase assembly and activation in PMNs, 78 and after uptake inhibits processing and activation of caspases-9, -8 and -3. Moreover, PS externalization and DNA fragmentation are significantly diminished and delayed, as is cell progression to an apoptotic morphology. 20 In this regard, F. tularensis is similar to A. phagocytophilum, as both pathogens not only prevent PICD but also profoundly inhibit constitutive PMN turnover as indicated by comparison to uninfected controls.
The data support a model whereby blockade of ROS production and downregulation of Bax mRNA and protein prevent PICD during tularemia. 79 At the same time, F. tularensis significantly alters the expression of more than 350 neutrophil genes directly linked to apoptosis and cell survival. 79 Although much remains to be determined, at a minimum, the underlying molecular mechanisms appear to include effects on the calpastatin-calpain-XIAP pathway (Fig. 1) as expression of CAST (which encodes calpastatin) is enhanced, and calpain-dependent degradation of XIAP is nearly ablated for at least the first 48 hours after infection. 79 Although cIAPs and CDKs are also upregulated by F. tularensis, the infected cells do not secrete IL-8, and other regulatory factors such as PCNA are downregulated.20,79 The bacterial factors that alter PMN lifespan remain obscure; however, a role for secreted factors, but not LPS or capsular polysaccharides, is established. 20
Viruses are perhaps the best examples of microbes with a vested interest in maintaining host cell viability, as they require host cell machinery to replicate. To this end, human cytomegalovirus inhibits neutrophil death by directly blocking caspase-8 activity, thereby preventing Fas-mediated apoptosis and preserving its replicative niche. 80 In contrast, respiratory syncytial virus enhances PI3K signaling and NFkB activation to increase expression of the pro-survival factor Mcl-1. 81
Pathogen acceleration of apoptosis and induction of cell lysis
Enhanced neutrophil apoptosis in HIV infection is believed to contribute to the elevated risk of secondary infections that are characteristic of AIDS. 82 Influenza A virus also increases apoptosis by a process that is ROS-dependent and includes enhanced expression of both Fas and Fas ligand. 83 At the same time, several bacterial pathogens either accelerate PMN apoptosis above and beyond what is typical of PICD or induce cell lysis to evade intracellular killing and return to the extracellular milieu. For example, the combined effect of accelerated PMN apoptosis and impaired efferocytosis enhances PMN progression to secondary necrosis and favors survival of Pseudomonas aeruginosa.84,85 In addition, other factors released from dying cells, such as elastase, contribute to disruption of the lung epithelium during cystic fibrosis. 86 Although the underlying mechanism is not entirely understood, the type III secretion system 87 and the toxic metabolite pyocyanin88,89 are essential for rapid PMN death. Other bacterial pathogens that accelerate PICD include S. pyogenes 13 and Streptococcus pneumoniae, 90 whereas intracellular Staphylococcus aureus trigger rapid necrotic lysis of neutrophils by a mechanism that does not involve the Panton-Valentine leukocidin.14,91
Neutrophils as Trojan horses
Although it has been known for several years that Leishmania promastigotes infect macrophages, differentiate into amastigotes, and replicate in lysosome-like compartments, recent data suggest that neutrophils may be the first cell type infected. Thereafter, PMN apoptosis is delayed in parallel with sustained mitochondrial integrity and upregulation of A1. 92 Moreover, it appears that the parasite may harness dying apoptotic neutrophils as vehicles for silent infection of macrophages following efferocytosis. 93 In the past few years, the concept of neutrophils as Trojan horses for infection of macrophages has been extended to include Yersinia pestis, 94 C. pneumoniae, 95 Toxoplasma gondii, 96 and Coxiella burnetii. 97 An emerging theme from these studies is enhanced disease progression via manipulation of macrophage phenotype and function in a manner that is not achieved by direct pathogen infection of this cell type.
Neutrophil Apoptosis in Inflammatory Disease
Delayed neutrophil apoptosis contributes to the pathology of many inflammatory and autoimmune diseases. This is perhaps best exemplified by the phenotype of persons with CGD who are at risk of life-threatening infections because of inherited defects in the phagocyte NADPH oxidase, yet also develop granulomas that obstruct the liver, lungs, and gastrointestinal tract secondary to defects in PMN turnover and clearance.54,98 Neutrophils from patients with acute arterial occlusions also exhibit delayed apoptosis, 99 and the number of circulating neutrophils correlates with mortality subsequent to cardiovascular events. 100 A recent study attributes this effect to the platelet-derived factor PF4, 101 identifying platelets as yet another source of neutrophil apoptosis modulation. Neutrophil turnover is also impaired in coronary artery disease, chronic obstructive pulmonary disease, and acute respiratory distress syndrome, but the underlying molecular mechanisms are not well defined.
In rheumatoid arthritis102,103 and anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis, 104 it is hypothesized that an accumulation of neutrophils contributes to the presentation and persistence of autoantigens. In agreement with this, disease severity correlates with exceptionally high numbers of apoptotic neutrophils in circulation. 105 At the molecular level, caspase expression is enhanced, whereas IAP expression is diminished, 106 and the robust accumulation of apoptotic PMNs combined with defective efferocytosis 107 increases release of DNA and other autoantigens. In contrast, the leukotriene LTB4 is elevated in psoriasis, cystic fibrosis, asthma, rheumatoid arthritis, gout, and irritable bowel disease, 108 and has been shown to delay neutrophil apoptosis through ROS-dependent degradation of the proapoptotic factor Bad. 109 Altogether, these data lend further support to the notion that delayed neutrophil apoptosis contributes to the pathology of chronic inflammatory disorders.
Therapeutic Intervention
Recently developed treatments for infections and inflammatory disorders exploit the biology of neutrophil apoptosis to drive resolution of inflammation and minimize tissue damage. For example, CDK inhibitors enhance apoptosis and decrease inflammation in animal models of arthritis, bleomycin- induced lung injury, pleurisy, and brain damage associated with pneumococcal meningitis.43,44 At the molecular level, enhanced apoptosis requires caspases but not NFkB or ERK, and is characterized by diminished expression of Mcl-1.43,110 Of particular interest is the CDK inhibitor R-roscovitine that can overcome the effects of pro-survival stimuli such as GM-CSF, TNFα, and LPS.43,110
Several treatments designed to block PMN survival signaling have also shown promise in animal models. Both NFkB and ERK inhibitors are able to enhance neutrophil apoptosis and decrease the number of inflammatory cells in murine models of pleurisy.63,111,112 Interestingly, Maiuri et al developed an NFkB decoy oligonucleotide that not only enhances neutrophil apoptosis but also increases their clearance by macrophages. 113 Although these treatments hold promise for inflammatory conditions, questions regarding their possible effects on other cell types remain to be answered before use in humans. On the other hand, TRAIL was recently demonstrated to induce apoptosis in models of zymosan-induced peritonitis and LPS-induced lung injury without any apparent effects on other cell types.114,115
Exploiting the inherently anti-inflammatory nature of apoptotic neutrophils, Ren et al demonstrated that infusion of apoptotic neutrophils enhances survival in a murine model of LPS-induced septic shock. 116 In addition to cytokine release and other pro-resolving effects, studies suggest that apoptotic neutrophils may also contribute to survival by acting as a “sink” to remove LPS molecules in the bloodstream. 116
Conclusions
Recent advances in the understanding of neutrophil biology have shed light on the mechanisms that control cell death and survival under homeostatic conditions as well as during infection and inflammation. The recent characterization of the PICD pathway and the discovery of neutrophil-specific regulatory factors such as PCNA underscore the unique features of this short-lived leukocyte. Although much remains to be determined, these findings together with our expanded understanding of the mechanisms of apoptosis manipulation by bacterial, fungal, parasitic and viral pathogens advances studies of infectious diseases, while also suggesting possible points of therapeutic intervention that may enable the development of novel treatments for inflammatory diseases.
Footnotes
Author Contributions
Wrote the first draft of the manuscript: JMM. Contributed to the writing of the manuscript: LAHA. Agree with manuscript results and conclusions: JMM and LAHA. Jointly developed the structure and arguments for the paper: JMM and LAHA. Made critical revisions and approved final version: LAHA. All authors reviewed and approved of the final manuscript.