
Abstract
Chloroplasts are organelles found in cells of green plants and eukaryotic algae that conduct photosynthesis. Knowing a protein's subchloroplast location provides in-depth insights about the protein's function and the microenvironment where it interacts with other molecules. In this paper, we present BS-KNN, a bit-score weighted K-nearest neighbor method for predicting proteins' subchloroplast locations. The method makes predictions based on the bit-score weighted Euclidean distance calculated from the composition of selected pseudo-amino acids. Our method achieved 76.4% overall accuracy in assigning proteins to 4 subchloroplast locations in cross-validation. When tested on an independent set that was not seen by the method during the training and feature selection, the method achieved a consistent overall accuracy of 76.0%. The method was also applied to predict subchloroplast locations of proteins in the chloroplast proteome and validated against proteins in Arabidopsis thaliana. The software and datasets of the proposed method are available at https://edisk.fandm.edu/jing.hu/bsknn/bsknn.html.
Keywords
Introduction
Chloroplasts are organelles found in cells of green plants and eukaryotic algae. They are believed to have originated from cyanobacteria through endosymbiosis. Chloroplasts play important functional roles in many biological processes such as photosynthesis and cellular metabolism. Similar to a cell that can be divided into several subcellular locations, the chloroplast is also subdivided into multiple subchloroplast locations. Knowing a protein's subchloroplast location information provides in-depth biological insights about the protein's roles in these biological processes.
Recent developments in high-throughput genome sequencing projects have resulted in an increasing number of raw chloroplast protein sequences stored in public databases. For the majority of these proteins, little knowledge is known about their subchloroplast locations. Therefore, computational methods that can predict subchloroplast localizations of chloroplast proteins are needed. However, despite the chloroplast proteome projects1–4 and various computational approaches5–8 to identify chloroplast proteins in proteomic scale, there are only a limited number of methods as to our knowledge for predicting protein subchloroplast locations. SubChlo 9 is the first method for predicting the subchloroplast locations of chloroplast proteins. The method is based on the evidence-theoretic K-nearest neighbor (ET-KNN) algorithm.10,11 Using pseudo-amino acid composition 12 as the feature set, the method achieved 67.2% overall accuracy in predicting proteins' subchloroplast locations on a dataset consisting of chloroplast proteins with less than 60% sequence similarities. ChloroRF 13 predicts subchloroplast locations using a feature vector of 531 physicochemical properties obtained from AAindex database. 14 Applying the Random Forest algorithm, ChloroRF achieved a comparable accuracy, 67.4%, as that of SubChlo on the same dataset. An extra benefit of ChloroRF is that it utilizes human-interpretable physicochemical properties, which can provide meaningful information for analyzing the mechanisms of protein subchloroplast localizations. Recently, a method called Subldent 15 was developed to identify submitochondria and subchloroplast locations. The method first created numerical series of hydrophobicity and polarity values from protein's amino acid sequence, and then applied a discrete wavelet transform to formulate them into a different representation of pseudo-amino acid composition. These features were then used to train a support vector machine classification model to predict protein's subchloroplast locations. For a complete list of subchloroplast localization methods and their details, please see Supplementary Table 1.
In this paper, we present BS-KNN, a bit-score weighted K-Nearest neighbor method for predicting proteins' subchloroplast locations. The method makes predictions based on a bit-score weighted Euclidean distance (BS-WED) computed from residue composition. For each subchloroplast location, it finds its K nearest neighbors (ie, proteins) to the query protein based on the BS-WED. Then the average BS-WED of the query protein to the K proteins is used as the distance of the query protein to this location. The query protein is then predicted to be in a subchloroplast location to which its distance is the smallest. The method achieved 68.0% overall accuracy in assigning proteins to four subchloroplast locations in cross-validation using amino acid composition. The method was then further improved by applying a heuristic feature selection process to choose pseudo-amino acid composition. The final method achieved 76.4% overall prediction accuracy in cross-validation. When tested on an independent set that was not seen by the method during the training and feature selection, the method still achieved 76.0% overall accuracy.
Materials and Methods
Dataset
We used the benchmark dataset that was used in the study of SubChlo, 9 ChloroRF 13 and ChloroRF. 15 The dataset can be downloaded from the website of SubChlo (http://bioinfo.au.tsinghua.edu.cn/subchlo/). The original dataset contained 736 proteins extracted from Swiss-Prot database release 56.2. 16 After discarding three proteins with incomplete Swiss-Prot IDs and removal of redundant proteins by CD-HIT 17 using a sequence similarity threshold of 60%, there were 253 proteins left. The final dataset (S60) contained 40 envelope proteins, 46 stroma proteins, 127 thylakoid membrane proteins and 40 thylakoid lumen proteins. The dataset used in this study was slightly different from the reported S60 datasets of SubChlo (262 proteins) and ChloroRF (261 proteins). This could be due to different versions of the CD-HIT program.
In most studies, a much stricter sequence similarity threshold (25% or 30%) was used to remove the redundancy among protein sequences. However, using such thresholds for this small dataset would greatly reduce the number of proteins in the final dataset. This would lead to insufficient training, and the performance evaluation would have little significance. Therefore we accepted a relaxed threshold (60%), which was also used in previous studies (ie, SubChlo, ChloroRF and Subldent). To show that our method does not suffer from generalization problems due to comparatively high sequence similarity among proteins in the dataset, we also compared our method with prediction based solely on similarity search.
Feature set
There were two feature sets investigated in this study, which were amino acid composition and pseudo-amino acid composition of protein sequences.
Amino acid Composition: The amino acid composition of a protein was calculated as:

Pseudo-Amino Acid Composition: The model of pseudo-amino acid composition was first proposed by Chou 12 to predict protein cellular attributes. Unlike the classical amino acid composition that consists of only 20 discrete numbers, the pseudo amino acid composition consists of 20+λ discrete numbers, among which the first 20 numbers represent the occurrence frequencies of 20 amino acids in a protein, and the remainders represent different ranks of sequence-order correlation factors. The model was then extended to include two sets of sequence-order correlation factors: delta-function set (λ discrete numbers) and hydrophobicity set (μ discrete numbers). The new model has been successfully applied to predict proteins' subcellular locations by Chou and Cai. 18
In this study, we investigated two sets of sequence-order correlation factors.
18
Suppose there is a protein X with a sequence of L amino acid residues: R1R2R3R4 … R
L
, where R1 represents the residue at sequence position 1, R2 the residue at position 2, and so on. The first set, delta-function set, consists of λ sequence-order-correlated factors, which are given by:


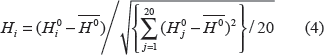
Bit-Score Weighted Euclidean Distance
For each query protein t, its distance to a training protein T is calculated as:

Bit-Score Weighted K-Nearest Neighbor Method
For each test protein, its BS-WED to every training protein in the training set was calculated. Then for each subchloroplast location, K shortest distances were chosen. For example, let Denv-1, Denv-2, …, D env-k be the K shortest distances between the test protein and proteins that locate at the envelope. Then, the distance between the test protein and the location of envelope was given by:
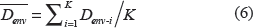
The distances between the test protein and every subchloroplast location were calculated separately. Then the test protein was assigned to a location to which its distance is the shortest.
Cross-validation, Independent Test, Self-consistency Test, and jackknife Test
The dataset was randomly split into 5 subsets. Four subsets (referred to as S60_A) were used to perform four-fold cross-validation and feature selection. In each round of experiments, three subsets were used as a training set, and the remaining subset was used as a test set. This procedure was repeated four times with each subset being used as a test set once. The overall performance was calculated. The fifth subset (independent set, and referred to as S60_B) served as the test set in the independent test stage, in which the classifier was trained using the four subsets (S60_A) and then tested on the independent set (S60_B). Note that the algorithm did not see the independent set during the feature selection stage and the training of the classifier.
Self-consistency test and jackknife test have been used by previous studies9,13,18,20 to evaluate the multiclass classification performance of protein localizations. In this study, we also evaluated the final BS-KNN method on S60 dataset using self-consistency test and jackknife test. In self-consistency test, proteins in the dataset were predicted using the classification model trained on the same dataset. Therefore, self-consistency test gives the most optimal estimation of the classification performance. In jackknife test, each protein in the dataset was used as the test protein once, and the remaining proteins were used to train the classifier. Therefore, jackknife test provides a more reliable estimation of the classification performance, especially when the dataset is small.
Performance Measurement
Performances were measured using accuracy/recall (RC) and precision (PR) for each subchloroplast location i:



Heuristic Feature Selection
We further extended the BS-KNN method by using pseudo-amino acid composition to calculate the BS-WED. However, not all the features were useful for the prediction of subchloroplast localizations. Also, some features might be correlated with each other, which could impair the prediction performance. We used a greedy feature selection method to select the most relevant features. The greedy search started with a feature set that included the composition of 20 amino acids. Let n be the size of the feature set. Then n = 20 at the beginning. The algorithm was divided into three stages: reduction, growth_I and growth_II. In the reduction stage, the size of the feature set was gradually reduced. First, one amino acid was removed, and the composition of the remaining n-1 amino acids was used to calculate the BS-WED. Four-fold cross-validation was used to evaluate the performance of the method by optimizing the overall accuracy. This step was repeated n times, so that every combination of n-1 amino acids was tried. The combination that improved the performance most was chosen. Thus, the size of the feature set was reduced from n to n-1. This reduction process was continued until removing any amino acid from the feature set would reduce the performance. At the end of the reduction stage, we reached a feature set that included the composition of N amino acids (N ≤ 20). Next, we entered the growth_I stage to increase the size of the feature set by adding the delta-function factors. One delta-function factor was temporarily added into the feature set, and the resulting feature set was used to calculate BS-WED. Four-fold cross-validation was used to evaluate the performance of the method. This step was repeated λ (ie, 20) times, so that every delta-function factor was tested. The delta-function factor that yielded the greatest improvement in performance was chosen and added to the feature set. Thus, the size of feature set was increased to N+1. The growth_I stage for including delta-function factor was continued until adding more delta-function factor would decrease the performance. Then we entered the growthII stage for including hydrophilicity factors. The growthII stage was processed similarly to the growth_I stage.
Results
Prediction Performance using only Amino Acid Composition
The BS-WED was developed as the distance measurement in the proposed BS-KNN algorithm. Only the composition of 20 amino acids was used to calculate the distance between the test protein and training proteins. Four-fold cross-validation was used to evaluate the performance. Various K values ranging from 1 to 15 were tried. As can be seen from Figure 1, the best performance was achieved when K 2. The proposed BS-WED achieved 68.0% overall accuracy in assigning proteins' subchloroplast locations. Notice that this had already outperformed the prediction performance of SubChlo (67.2%) and ChloroRF (67.4%). For comparison purposes, we also showed the prediction performances of the K-NN method using standard Euclidean distance. As can be seen from Figure 1, the BS-WED developed in this study is a better distance measurement than standard Euclidean distance in predicting subchloroplast locations for K values from 1 to 15.
Prediction accuracies of K-NN for various K values (1-15) based on Euclidean distance (ED) vs. bit-score weighted Euclidean distance (BS-WED).
Prediction Performance using Selected Pseudo-Amino Acid Composition
The performance of BS-KNN based on selected pseudo-amino acid composition.

The performance of BS-KNN using selected pseudo-amino acid composition on S60 dataset by self-consistency test and jackknife test.

Comparison with Prediction Solely Based on Similarity Search
Similarity searches have been widely used to infer protein functions. If two proteins are highly similar in sequence, then they might share similar functions, structures, and evolutionary origin. For each test protein, we conducted a homologous search on the training set using the BLAST program. 19 The test protein was predicted to be at the same location (ie, thylakoid lumen, stroma, thylakoid membrane, or envelope) as that of the most homologous protein. Using the same dataset partition, the similarity search only achieved 65.0% overall accuracy when evaluated by four-fold cross-valuation, which was much lower than the proposed BS-KNN method.
Comparison with Previously Published Methods
Comparison of BS-KNN with previously published subchloroplast localization methods on S60 dataset by jackknife test.

Proteome scan
The proposed BS-KNN method was also applied to scan the chloroplast proteomes downloaded from plprot. 21 The plprot contains 690 chloroplast proteins of Arabidopsis thaliana as of March 2011, among which 258 (37.4%) proteins were predicted to be in the stroma, 139 (20.1%) proteins were predicted to be in the envelope, 99 (14.3%) proteins were predicted to be in the thylakoid lumen, and 194 (28.1%) proteins were predicted to be in the thylakoid membrane. Though some of these predictions would need to be validated, they could provide suggestive information to the future chloroplast proteome projects.
In this paper, we also validate our method against proteins in Arabidopsis thaliana downloaded from PPDB (http://ppdb.tc.cornell.edu/default.aspx), a Plant Proteome DataBase for Arabidopsis thaliana and maize (Zea mays). The extracted proteins with annotation of subchloroplast locations of envelope, stroma, thylakoid lumen side (lumen), and thylakoid-integral (thylakoid membrane) used in this study were based on their subcellular proteomes data set. There were 958 proteins in total, of which 345 (36.0%) proteins were predicted to be in the stroma, 208 (21.7%) proteins were predicted to be in the envelope, 127 (13.2%) were predicted to be in the thylakoid lumen, and 278 (29.0%) proteins predicted to be in the thylakoid membrane. In total, 52.9% of these proteins (experimental verified or predicted by previous studies) have been correctly predicted. For comparison, we also applied SubChlo to predict proteins in Arabidopsis thaliana, and it achieved 49.3% accuracy.
The proteins in plprot and Arabidopsis thaliana and their prediction using BS-KNN can be found at https://edisk.fandm.edu/jing.hu/bsknn/genomeScan/.
Discussion
In this paper, we present a BS-KNN algorithm for predicting protein subchloroplast locations. For each query protein, BS-WED was used as the distance measurement to find its K nearest neighbors from each location. To further improve the method's prediction performance, we investigated the pseudo-amino acid composition. Compared with the classical amino acid composition, the pseudo-amino acid composition provided more sequential and physicochemical information at different orders. By applying a heuristic feature selection process, the final method achieved 76.4% overall accuracy in classifying proteins into 4 subchloroplast locations using selected pseudo-amino acid composition by four-fold cross-validation. When evaluated on an independent test dataset, the method achieved a consistent accuracy of 76.0%. The method also achieved 75.9% overall accuracy by jackknife test. This shows that our method does not suffer from generalization problem and it has consistent prediction performance. We also applied our method to annotate proteins in the chloroplast proteome and validated the method against proteins in Arabidopsis thaliana.
The proposed BS-KNN method used a bit-score weighted Euclidean distance (ie,
In conclusion, the proposed bit-score weighted K-nearest neighbor algorithm is an effective method for predicting the subchloroplat location of proteins.
Supplementary Table
List of computational methods for protein subchloroplast localization.

Footnotes
Acknowledgments
The project is partially supported by the grant from Howard Hughes Medical Institute awarded to Franklin & Marshall College.
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.