
Abstract
The subtree prune and regraft distance (
Introduction
Phylogenetic trees are used to describe evolutionary relationships. DNA or protein sequences are associated with the leaves of the tree and the internal nodes correspond to speciation or gene duplication events. In order to model ancestor-descendant relationships on the tree, a direction must be associated with its edges by assigning a root. Often, insufficient information exists to determine the root and the tree is left unrooted. Unrooted trees still provide a notion of evolutionary relationship between organisms even if the direction of descent remains unknown.
The phylogenetic tree representation has recently come under scrutiny with critics claiming that it is too simple to properly model microbial evolution, particularly in the presence of lateral gene transfer (LGT) events (Doolittle, 1999). A LGT is the transfer of genetic material between species by means other than inheritance and thus cannot be represented in a tree as it would create a cycle. The prevalence of LGT events in microbial evolution can, however, still be studied using phylogenetic trees. Given a pair of trees describing the same sets of species, each constructed using different sets of genes, a LGT event corresponds to a displacement of a common subtree, referred to as a SPR operation. The SPR distance is the minimum number of SPR operations, denoted by
Tree bisection and reconnection (TBR) is a generalization of SPR that allows the pruned subtree to be rerooted before being regrafted. Computation of the TBR distance (
SPR Distance Computation is NP-Hard for Unrooted Trees
Hein et al. (1996) showed that computing the size of a the Maximum Agreement Forest (MAF) of two trees is NP-Hard by reducing it from Exact Cover of 3-Sets (X3C). Later, Allen and Steel (2001) proved that this result is insufficient to show the hardness of unrooted SPR distance because there is no direct relationship between MAF size and
Definition 2.1
An
Definition 2.2
The
We now review the polynomial-time reduction from X3C to MAF provided by Hein et al. (1996), clarifying their notation to reflect that each element of

Reduction of an instance of X3C to |
Tree
(Hein et al. 1996) show that
Proving that
Cut leaves
Cut the leaves
We now show that given two trees
Prune leaves
Prune the leaves
There is a one-to-one correspondence between cuts formed when creating the MAF and SPR operations that can transform
Definitions
All trees referred to in this paper, unless otherwise stated, are unrooted binary phylogenetic trees. Such trees have interior vertices of degree 3 and uniquely labeled leaves. Given a tree

2(a) Original tree. 2(b) Edge
The reduction rules referred to above only serve to transform the original problem into smaller subproblems. These subproblems must still be solved with an exhaustive search as the problem is NP-Hard (see proof in Appendix). Let
While it is still an open problem to determine if reduction rules can be found to reduce

Heuristic improvements
A subtree reduction replaces any pendant subtree that occurs in both input trees by a single leaf with a new label in each tree as as shown in Figure 3(a). A chain reduction, illustrated in 3(b), replaces any chain of pendant subtrees that occur identically in 

Reduction rules applied to a tree. 3(a) A subtree is reduced to a leaf. 3(b) A chain of length
In addition to applying reductions on the input trees, intermediate trees visited during the breadth-first search can be likewise reduced. For example, if
Because the number of trees visited in each iteration of the exhaustive search increases exponentially, the asymptotic complexity is bounded by the number of trees explored in the final iteration. It follows that the order in which these trees are searched can have a critical impact on the running time. We attempt to increase the probability that the tree upon which the search is completed is visited near the beginning of an iteration by sorting the trees in each iteration according to how many leaves are eliminated in by subtree reduction. Our hypothesis is that trees with larger common subtrees are more likely to be near the destination tree. Since at most

A cluster is the leaf set of a pendant subtree.

Example of trees whose common clusters cannot be maintained by a minimal SPR path.
Datasets
The datasets were chosen to analyze the merits of the heuristics discussed in the previous section as well as evaluate our algorithm in a realistic setting. To these ends, we bench-marked our algorithm on a variety of randomly generated trees, as well as trees created by Beiko et al. (2005) in the course of analyzing the proteins from the 144 sequenced prokaryotic genomes available at the time. Two sets of random trees were generated, one by the Yule-Harding model and the other by random walks. Yule-Harding trees are constructed by first creating an edge between two randomly selected leaves, then randomly attaching the remaining leaves to the tree until none are left. The random walk dataset consists of pairs of trees such that one of which is generated by the Yule-Harding model and the other is created from the first by applying a sequence of between 2 and 8 random SPR operations (Beiko and Hamilton, 2006). The size of the datasets, along with the average distances computed by our algorithm are presented in Figure 5. In some cases, the program ran out of memory before finding the solution. The fraction of instances successfully resolved for each type of data is listed in the “% Resolved” column (Fig. 5(a), 5(c) and 5(e)).
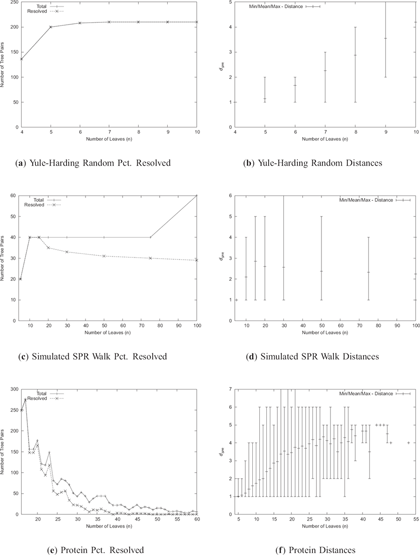
Size, success rate and distance distributions for each dataset. For the protein data, no trees of size greater than 60 were resolved.
The algorithm described in Section 3 was implemented in C++ and benchmarked on a 2.6Ghz Pentium Xeon System with 3G of RAM. The source code is available at http://morticia.cs.dal.ca/lab_public/?Download. This program was executed for all pairs of trees described in Figure 5 with and without the various heuristic optimizations discussed previously. Graphs 6(a), 6(c) and 6(e) in Figure 6 display the effectiveness of the reduction rules’ ability to reduce the input trees. As could be expected, the trees in the protein and random SPR walk datasets are reduced more than the two random datasets as their ratios of size to distance are much higher. In all cases, the amount of reduction increases in correlation to the mean distance rather than

Experimental evaluation of the different heuristics on the three datasets. The effect of the reduction rules on the input tree sizes is displayed on the left. The improvements to the running time made by reducing and sorting intermediate trees are displayed on the right.
The performance of the remaining heuristics is displayed in terms of running time in graphs 6(b), 6(d) and 6(f) in Figure 6. Applying the reductions to intermediate trees provided very little performance gain, implying that the search space is dominated by trees with few common subtrees and chains. However, sorting the trees visited in each iteration of the search by the number of leaves reduced had a significant impact on the running time for all of the harder cases (
The computation of SPR distances between unrooted phylogenetic trees can be used to compare the evolutionary histories of different genes and provide a lower bound on the number of lateral transfers. Little previous work has been done on this problem though many related tree metrics have been relatively well studied in the literature. The reason for this appears to be primarily due to less insight into the problem's structure (no known MAF reduction) rather than lack of interest. In this paper we revisited the problem of unrooted SPR distance, showing that it is NP-Hard and providing an optimized algorithm and implementation to solve it exactly. The algorithm is based on dividing the problem into two searches and making use of heuristics such as subtree reductions and reordering. This algorithm was able to quickly compute the exact distance between the majority of proteins belonging to 144 available sequenced prokaryotic genomes and their supertree. Our method can also be used to improve the brute force search component of TBR and rooted SPR distance computation.
Though a polynomial time solution is unlikely due to its NP-Hardness, some possible avenues of future work on this problem remain. One is to show that chain reductions do not affect the distance, a conjecture that is supported by our experimental results but for which an analytical proof remains absent. This result would be sufficient to show that unrooted SPR distance is fixed parameter tractable, being exponential only in terms of the distance and not the size of the trees. Bordewich et al. (2007) used a decomposition by common clusters was used with significant practical success. We showed that such a technique cannot be directly applied to the problem of unrooted SPR distances but perhaps a variation of this technique can.
The contributions of this paper can thus be summarized as follows: (1) We show that SPR distance computation is NP-hard for unrooted trees. (2) We present an efficient heuristic algorithm for this problem and benchmark it on a variety of synthetic datasets. Our algorithm computes the
Footnotes
Acknowledgment
This research partially supported by the Natural Sciences and Engineering Research Council of Canada and Genome Atlantic.