
Abstract
Homology detection has evolved over the time from heavy algorithms based on dynamic programming approaches to lightweight alternatives based on different heuristic models. However, the main problem with these algorithms is that they use complex statistical models, which makes it difficult to achieve a relevant speedup and find exact matches with the original results. Thus, their acceleration is essential. The aim of this article was to prefilter a sequence database. To make this work, we have implemented a groundbreaking heuristic model based on NVIDIA's graphics processing units (GPUs) and multicore processors. Depending on the sensitivity settings, this makes it possible to quickly reduce the sequence database by factors between 50% and 95%, while rejecting no significant sequences. Furthermore, this prefiltering application can be used together with multiple homology detection algorithms as a part of a next-generation sequencing system. Extensive performance and accuracy tests have been carried out in the Spanish National Centre for Biotechnology (NCB). The results show that GPU hardware can accelerate the execution times of former homology detection applications, such as National Centre for Biotechnology Information (NCBI), Basic Local Alignment Search Tool for Proteins (BLASTP), up to a factor of 4.
KEY POINTS
Owing to the increasing size of the current sequence datasets, filtering approach and high-performance computing (HPC) techniques are the best solution to process all these information in acceptable processing times.
Graphics processing unit cards and their corresponding programming models are good options to carry out these processing methods.
Combination of filtration models with HPC techniques is able to offer new levels of performance and accuracy in homology detection algorithms such as National Centre for Biotechnology Information Basic Local Alignment Search Tool.
Keywords
Introduction
Over the last few years, homology detection algorithms have evolved rapidly in terms of accuracy and performance. Historically, the first relevant milestone in this collection is the Smith–Waterman (SW) algorithm. 1 By using exhaustive dynamic programming (DP) techniques, 2 this algorithm calculates the similarity between two amino acid sequences by a local gapped alignment and yields the optimally scoring path in terms of the given model. However, their main drawback is that they are very computationally expensive, especially when comparing large sequences. Heuristic algorithms, such as Basic Local Alignment Search Tool (BLAST) 3 and FASTA, 4 have been developed to detect homologies in large sequence databases. These statistical models can obtain alignments much more rapidly and often closely match alignments that would be generated by DP methods.
As the number of sequences keeps increasing much faster than our ability to analyze them, the need for accelerating homology detection algorithm is more than ever a central problem in computational biology. Based on the idea of accelerating these algorithms, several high-performance computing (HPC) hardware have been used to date.
Field-programmable gate array (FPGA) is the first component in this collection. By profiling the source code and accelerating the most heavily used modules, algorithms such as BLAST5–8 and SW9–11 have been accelerated with significant speedups. However, FPGA has some hardware limitations. Because the code size is translated into a limited chip area, it reduces the number of improved application modules. Furthermore, due to the high costs involved, it is not able to achieve a good cost-effective performance.
Graphics processing units (GPUs) are the second option and the most feasible alternative to FPGA. By refactoring the original source code, algorithms such as BLAST12–14 and SW15–17 have been accelerated. In contrast to FPGA, there is no hardware limitation for GPU, but the computational complexity of heuristic models in homology detection algorithms, the handling of the different GPU memories (ie, shared or global), and their long latency periods make it challenging to achieve a good cost-effective performance.
Currently, there is a new line of research based on detecting remote homology proteins with predictive techniques.44–47 By using multiple kernel learning models, such as support vector machines and learning to rank, all evaluated solutions are able to help homology detection algorithms or prefiltering approaches in their worst-case scenario, ie, very low sequence similarity (<30%). Despite the fact that these learning methods have a significant computational overhead, HPC hardware (ie, GPU or FPGA) can help to reduce this bad performance effect.
Finally, there is an alternative approach based on prefiltering amino acid sequences. The main idea is to quickly reduce the number of database sequences to a small fraction by removing those sequences considered irrelevant according to a given theoretical model.18,19 Our proposed application
20
falls into this category and differs from previous ones in that it exploits the massively parallel processing power of GPU hardware. Such an approach is really innovative because it can be used with any homology detection algorithm and performs daily genomic studies in less time and more accurately depending on the algorithm used. Following this reasoning and according to recent studies,
21
the National Center for Biotechnology Information (NCBI) BLAST accuracy, in terms of search space, is inversely proportional to the number of sequences into the database. It is a very common programming technique to avoid long run execution times for big input data. The work proposed in this article uses this novel filtering approach in conjunction with GPU hardware. The primary result is a GPU-accelerated NCBI BLASTP that achieves a very similar result to the original, with 95% accuracy evaluating 20,000 query sequences
20
from different genomes, skewed or not, and a factor of 4× improvement in performance. Furthermore, since the prefiltering method is completely independent of the algorithm search space, users may keep the extra output at no cost and impact to performance. Following is a summary of the most important points of this work:
The prefiltering application requires only an off-the-shelf GPU hardware; it is likely to be cost-effective and could achieve a widespread use. The proposed implementation is based on heuristics and can be replaced by another filter or statistical model. The prefiltering method is completely transparent and fully compatible with any homology detection algorithm, such as NCBI BLASTP.
All the source code, documentation, and installation instructions are freely available in the GitHub repository 42 under MIT license.
The remainder of this article is organized as follows. The Background section provides an overview to the reference homology detection algorithm, NCBI BLASTP, the HPC techniques used with NVIDIA's GPU and some theoretical assumptions about the proposed architecture. The Related works section explains some of the most relevant approaches that have been investigated to parallelize NCBI BLASTP. The Implementation section describes the inner details of the filter and its HPC assumptions relative to the original algorithm, and finally, the Evaluation section evaluates the performance and accuracy of the proposed implementation with different examples and presents some conclusions about the research work carried out in the Conclusions section.
Background
NCBI BLAST
Homology between two amino acid sequences is a bidirectional relationship, ie, alignment, between their characters without reordering, but with the possibility of insertions or deletions. This relationship results into an alignment score that is determined a priori by biological significance. By using DP techniques, the highest scoring alignment between a query sequence of length m and a database sequence of length n can be found in time O(mn). However, heuristic algorithms, eg, NCBI BLAST, have partially ignored these kinds of techniques due to the fact that they result inefficient in terms of performance when used with large databases.
Homology detection applications based on heuristic methods are the starting point of most of the HPC genomic research. These investigations aim to reduce the execution times and increase the accuracy of the algorithms. In particular, due to the extended use in different biological fields, our research has selected NCBI BLAST as the reference algorithm.
NCBI BLAST is one of the most known and used bioinformatic applications. This algorithm is highly parallelizable
22
and is mainly based on statistical methods with hit-and-extend heuristics for proteins and DNA. When executing, it goes through the following steps:
Seeding: identifying high-score matches. Extension: extending high-score matches and merging nearby extensions. Evaluation: evaluating the obtained extensions and calculating high-scoring segment pair (HSP) alignments.
As a preliminary phase to stepping up the original algorithm, current versions of NCBI BLAST try to filter low-complexity regions. The problem surrounding these regions is that they might give high-score values that confuse NCBI BLAST to find significant sequences in the database. Algorithms such as SEG
23
for proteins and DUST for nucleotides are some examples of existing algorithms that might help to solve this problem. Then, NCBI BLAST is structured into the following steps:
For each k-letter, also know as lmer, in the query sequence (seeding):
Scan the database sequences for exact matches with the remaining high-scoring words (hitting). Extend the exact matches to HSP (extension):
Stretch a longer alignment between the query and the database sequence to the left and to the right directions and from the position where the exact match was found. List all the HSPs in the database whose score is higher than a specific user-defined cutoff score. Evaluate the significance of the HSP score (evaluation). This process evaluates the statistical significance of each HSP score by exploiting the Gumbel extreme value distribution and Karlin–Altschul equation. Make two or more HSP regions into a longer alignment.
Report every match whose expected score is lower than a user-defined threshold parameter.
General-purpose GPU architecture
GPU is a dedicated graphic rendering hardware, which can be found nowadays on every personal computer, smartphone and tablet. Thanks to its massively parallel architecture, a GPU can run trillions of instructions per second for both graphical and non-graphical applications. A GPU used for non-graphical applications is commonly known as general-purpose graphics processing unit (GP-GPU). The performance reached by GP-GPU has made this hardware a usual part of HPC clusters. In fact, some supercomputer vendors have included GPGPU inside their parallel compute blades. Cray XK7 48 supercomputer is an example with NVIDIA's GPUs.
There are many different GPU architectures and models. NVIDIA and AMD are the most popular GPU manufacturers. Multiple research and testing have been developed to evaluate which technology gives a higher performance.29,30 Given that NVIDIA's CUDA language provides, in general, greater control than other GPU languages, we have opted to use NVIDIA and its CUDA programming technology.
Typically, GPU devices are external to the microprocessor. Microprocessor and GPU connect and communicate through Peripheral Component Interconnect Express. This poses that a memory copy from the host to the GPU has to be done before using the host data. This fact can make a GPU very inefficient if the data copy takes much time compared to the processing time. The NVIDIA's GPU architecture consists of a large number of cores of stream processors (SPs), grouped into stream multiprocessors (SMs). The SPs are small processors able to perform integer operations and simple-precision operations. The SM also contains double-floating point units, registers, level 1 cache, and a shared memory. Each SM shares these resources among its SP cores. In a similar way, every SM shares a level 2 cache and the global memory between the others SMs. Furthermore, this technology is continually evolved. For example, the NVIDIA's Fermi architecture provides up to 16 SMs, each one with 32 SP cores
31
and the newer Kepler architecture provides up to 15 SMs, each one with 192 SP cores and 64 double-precision units.
32
Figure 1 shows an overall design of a NVIDIA's GPU architecture.
NVIDIA GPU general architecture.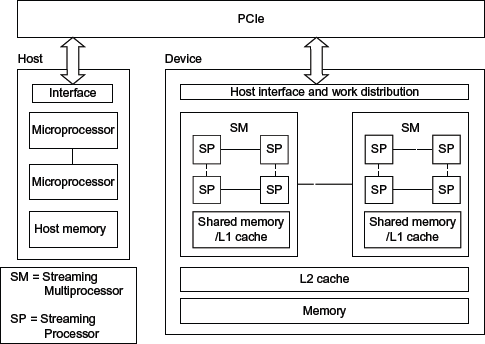
The CUDA programming model provides the possibility of programming parallel functions that will be executed on the GPU. Each parallel function is called CUDA kernel. Each kernel is a part of the application code, usually written in ANSI C, which can be executed in parallel with other kernels. The number of parallel executions depends on the required resources by the CUDA application and how many of them are currently available. Each kernel is launched on a grid. A grid is composed of a set of blocks, which can be defined as one, two, or three dimensional. In turn, each block is composed of a set of threads that can be also defined as one, two, or three dimensional. Each thread runs on an SP processor, and each block is executed on an SM. Owing to the architecture previously explained, different threads of the same block can share memory very efficiently and without having to access global memory.
To obtain a good performance, the programmer must ensure that the thread execution may not diverge in excess, as this would create serialization of execution between the threads of the same block. The programmer must keep in mind the total number of threads and their distribution between blocks. The programmer should also consider the amount of shared memory used by each thread and other possible architectural considerations. Figure 2 depicts how CUDA is organized. More information about CUDA programming model can be found in CUDA reference.
33
CUDA programming model.
The OpenCL programming model, as well as CUDA, is a low-level extension of C/C++ for heterogeneous computing that runs on CUDA-powered GPUs provided by Khronos OpenCL Working Group. 34 In the same way as CUDA, there is a sequential part of the application code, ie, kernel, that is executed by all individual threads. This part of the code is written using a limited subset of the ANSI C programming language on a GPU.
GPUs are specially well suited to address problems that can be expressed as data computations in which the same program is executed on many elements in parallel. Thus, to achieve a reasonable parallel efficiency, memory optimization schemes have to be adopted carefully to use the three layers of memory hierarchies: register, shared memory, and global memory.
Filtering sequences
According to previous studies,3,4 some heuristic models are based on filtration. As Equation 1 depicts, this filtering model is based on the Karlin-Altschul formula and its scoring scheme:
Obtain m value (query length) and n value (database sequence length). As Equation 2 shows, obtain H value for either ungapped or gapped alignment:
As Equation 3 shows, obtain the length adjustment variable for either ungapped or gapped alignment:
According to the obtained previous values and as Equation 4 shows, compute the effective search space:
As Equation 5 depicts, compute the raw score corresponding to the user-defined given threshold:
where Eval is a user-defined value (10.0 by default). Finally, the number of alignments expected is the value of Eval, and the smaller the Eval, the greater the significance alignment.





Related Works
As the Background section introduced, many approaches have been investigated to improve NCBI BLAST in the past with HPC or theoretical techniques.
On multi-core processing, the NCBI has implemented a version of BLAST by using POSIX threads, 49 also known as pthreads. This variant has the advantage that it can be used with commodity hardware in most cases, but according to Amdahl's law, once a certain number of cores are met, it becomes a high-cost hardware solution.
From cluster perspective, there are some parallel implementations, such as MPIBLAST, 35 used into heterogeneous cluster platforms, also known as Beowulf cluster. Despite the fact that the speedup tendency growth is in line with the number of cluster processors, the I/O bottlenecks between cluster nodes and the horizontal scalability in terms of number of processors become a high-cost hardware solution.
FPGA variants36,43 provide a high-cost hardware solutions with a significant performance improvement as much as 376-fold. However, according to the Background section, the number of improved application modules depends on the chip area. This limitation, together with the fact that FPGA has a very high price tag, means that these solutions become unaffordable.
GPU represents a balanced solution between cost and performance. Our research and proposed implementation has focused on parallelizing BLAST with GPU hardware. At this point, either Liu et al. 13 with CUDA-BLASTP, Vouzis and Sahinidis 14 with GPU-BLAST, or Xiao et al. 12 are based on the parallelization of all compute stages of NCBI BLAST. Following this reasoning, Liu et al. 13 with CUDA-BLASTP evaluated the performance of their proposed algorithm with only five query sequences, Vouzis and Sahinidis 14 with GPU-BLAST evaluated their corresponding performance with 51 query sequences, and Xiao et al. 12 used 1,000 query sequences without any kind of information about their phylogeny. In all cases, the number of query sequences and variety are far from sufficient to get a real performance perspective. By contrast, the proposed implementation does not develop a new version of NCBI BLAST and its compute stages to achieve a performance improvement, but it designs an ad hoc solution completely independent of future changes in NCBI BLAST. Furthermore, according to our previous work, 20 close to 20,000 query sequences from different genomes, skewed or not, and protein families have been evaluated in terms of performance and accuracy.
Implementation
The theoretical model, outlined in the previous section and described in detail in our previous work, 20 introduced the hit proximity concept. This approach is enough to establish a strong relationship between hits and the relevance of an alignment. This model has been designed and implemented according to a specific architecture, multi-core and multi-GPU schema, and is divided into two main elements, a lookup sequence table and a GPU filter implementation.
Database format
The first step in our implementation is to format the target sequence database. By using custom lookup tables and linked lists, this process aims to reduce the computational costs of hitting process to direct access O(1). In contrast to previous formatting systems with standard lists, such as NCBI formatdb tool, our proposed formatting model is based on basic hash functions and multi-core processing. This performance improvement concerns the inner details of lookup tables.
As we see in Figure 3, the proposed formatting system creates a specific set of threads. The total number of threads depends on the number of physical cores, without hyperthreading capabilities, within the computer. Each thread iterates over each sequence, splitting into lmers and processing the corresponding hash function. These calculations depend on the sequencing model, ie, proteins and nucleotides. Equation 6 shows the corresponding hash function for proteins.
Hashing database sequence architecture.
Once the hash index is obtained, each thread checks if the hash table entry has previous values. If so, it reallocates the memory space and inserts the sequence number into the corresponding linked list. The decision to use these data structures for each hash table entry is based on clarity, simplicity and fast prototyping grounds. All memory allocations are considered as atomic operations to avoid concurrence problems.
Owing to the fact that GLIBC hash table implementation
37
is very complex and hard to integrate, a custom model has been developed. This implementation is based on a bidimensional dynamic matrix and a custom hash function. As Equation 6 shows, these calculations depend on the lmer size. The usage of lmer or k-letter term is widely recognized in computational biology. It means the smallest unit in homology detection algorithms that represents a specific number of residues, usually 3-lmer for proteins to map directly with other reference applications, such as NCBI BLAST. Consequently, our proposed database model for proteins, with an alphabet size of 24 residues, would allocate 24
3
entries, ie, 55.296 bytes, in CPU memory.
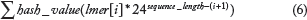
Finally, as a result of the formatting process, a binary database will be returned for further filtering analysis.
Filtering model
The filtering model, shown in Figure 4, consists of four compute stages. The processing policy consists of a preformatted database and a set of query sequences that are transferred to the GPU global memory. On the one hand, the preformatted database consists of a precalculated lookup table with database sequence information. On the other hand, the set of query sequences is a precalculated sequence stream with a variable size. Each query sequence is split into lmers and grouped into multiple blocks of threads, known as CUDA warps. Focusing on CUDA warp splitting implementation, zero padding has a great relevance because it is essential for avoiding divergence into the hitting policy.
GPU filtering model.
According to the upper section of Figure 4, each thread is responsible of hitting its assigned lmer. Such a process starts indexing the query sequence lmer according to the same hash function used in the formatting database section. Then, each thread writes 1 into the database stream if the corresponding lmer exists in the database sequence. Generally speaking, this database stream is to be understood as a bidimensional matrix where rows represent all database sequences and columns represent each different lmers.
Once all threads have finished their jobs and have been synchronized by using CUDA barriers, the two-step reduction process begins. The first step, shown in Figure 5, is based on all the threads working together in blocks, and according to the CUDA warp size, to obtain the sum of the items contained on their corresponding CUDA warp.
38
At this point, it is important to consider the division policy by CUDA warps because it is the only way to guarantee a full parallelization between threads and thus reduce the divergence between them.
CUDA reduction model.
Finally, once all existing CUDA warps have finished, the second step comes on the scene to obtain the minimum score or likelihood percentage. Then, it returns the evaluated sequence, in FASTA format, to CPU if it overcomes the bias introduced by parameter.
Additional considerations
There are some architectural considerations implemented to achieve the highest performance on NVIDIA's GPU and CUDA.
The first assumption is the processing policy. Data transfer overhead between GPU and CPU is one of the main bottlenecks in parallel applications and specially in those algorithms with huge data transfer.
39
In order to reduce the I/O overhead, the GPU global memory is split into two sections. While the first section has processing data, the second section is filled by using CUDA streams
40
and asynchronous memory transactions. This feature is available since NVIDIA's Fermi architecture development. Therefore and according to Figure 6, the proposed processing policy reduces the data transfer overhead to just the first transfer.
Processing policy comparison.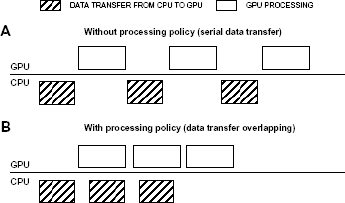
The second consideration into the model definition is the hitting policy. This policy involves two main problems: coalescence and divergence. On the one hand, to solve these coalescence problems, it is necessary to establish contiguous memory access by GPU threads and minimize the overhead caused by L1 and L2 cache memory misses. As Figure 4 shows, either memory access or cache memory misses are designed to reduce this problem. On the other hand, the minimization of divergence, ie, the effect caused by GPU threads finishing their assigned jobs in different execution times, has been achieved according to the hitting process and only those threads with zero padding data generate a little divergence into the global execution time.
The third consideration to take into account, specially for huge datasets, is the architectural decision to use global memory instead of shared, constant or texture memory. This decision is mainly due to a number of capacity planning constraints, such as memory size and CPU–GPU data transfer overhead.
Evaluation
This section validates the proposed implementation into two different ways: performance and accuracy. It completes our previous evaluation work 20 that evaluated the following genomes: Anaplasma marginale genome with 9,000 sequences, Escherichia coli genome with 5,000 sequences, Buchnera aphidicola genome with 1,000 sequences, and Pseudomonas putida genome with 1,000 sequences. These genomes were executed with non-redundant database from NCBI.
The accuracy test consists of a fine-grained validation procedure that aims to determine that the obtained results are correct. Once this validation is obtained, we can certify the compatibility of the proposed methodology with other reference applications. As a result, these tests conclude with high accuracy levels in the experiments performed:
Between 70% and 80% for sequence exact matches. Close to 100% for inclusion of sequences.
On the other hand, performance tests are part of the second stage of this evaluation process, which aims to mix the obtained accuracy levels in the previous phase with good performance in terms of execution time. These tests have been implemented with the NVIDIA's GPU card NVIDIA Tesla K40c. This card is a single GPU from Kepler architecture with 876 MHz of GPU clock rate, 12 GB GDDR5 device memory, and 2,880 CUDA cores.
This GPU card is installed into an HPC hardware architecture with an Intel Xeon E5-2630 processor, PCI Express 3.0 connections, and 31 GB RAM. This processor is composed of six non-uniform memory access nodes with 12 physical cores and hyperthreading enabled, ie, 24 real cores visible for the operating system. Furthermore, these cards run into a GNU/Linux Fedora 18 (x86_64) system with kernel version 3.11.10-100, CUDA version and runtime 6.0, and CUDA compute capability 3.5.
The sequence database used for these tests is the full GenBank Nonredundant Protein Database, which contains 3,163,461,953 amino acids in 9,230,955 sequences. The comparison between query datasets and sequence database has focused into two different families, polyprotein viruses and proteobacteria. The reason for selecting these families is because their sequences has appropriate sizes, ie, long lengths, to carry out the worst-case scenarios for GPU memory management.
Performance comparison.

Filtering comparison.

However, according to Amdahl's law, the proposed model performance is directly related to the likelihood filter threshold. This input parameter determines the number of sequences that the reference application will analyze. In particular, the evaluation stage is divided into two different scenarios. On the one hand, best-case scenarios define a high likelihood filtering threshold, ie, 95%, discarding a minimum of 90% of subject database sequences and obtaining a 4× speedup. On the other hand, worst-case scenarios define a low likelihood filtering threshold, ie, 60%, discarding a minimum of 50% of subject database and obtaining a 2× speedup.
During these tests, the average temperature of both GPUs was never >55 °C. According to Hong and Kim, 41 the obtained value followed a standard value. It demonstrates that the proposed implementation can achieve energy savings by using a minimal number of GPU cores and a less intensive utilization.
Future works are focused on conducting the same kind of tests with NVIDIA's GTX Titan GPU card. This card is a single GPU from Kepler architecture with 837 MHz of GPU clock rate, 6 GB GDDR5 device memory, and 2,688 CUDA cores. Our assumption is that this kind of GPU cards could provide similar performance results as NVIDIA Tesla K40c but with a significant budgetary savings. Unlike other related research works, this comparison can provide an added value to the current research, offering a different perspective in terms of commodity hardware and HPC in biomedical and biological scenarios.
Conclusions
Next-generation sequencing systems are revolutionizing homology detection algorithms such as NCBI BLAST. Novel statistical and numerical methodologies have been implemented for improving these algorithms in terms of performance and accuracy.
The highly parallelizable architecture of NVIDIA's GPU and CUDA has achieved a massive sequence analysis with low cost using commodity hardware and improvements up to four times faster than standard algorithms.
Unlike other existing solutions based on GPU or FPGA,12–14 the proposed implementation is completely independent of the original algorithm because it is not based on a new implementation of all compute stages. As a result, the proposed ad hoc solution gives a significant flexibility because it can be connected to new releases of the reference algorithm or even with other similar applications.
Several sets of experiments, from different protein families, have been conducted to evaluate and validate the proposed filtering model. As a conclusion, this approach can reduce execution times of sequencing algorithms using commodity hardware and, depending on the algorithm used, improve their accuracy.
Biography
Germán Retamosa received his M.Sc. degree in Computer Science and Telecommunications from Universidad Autónoma de Madrid, Spain, in 2009 and is currently finishing his Ph.D. in Computer Science and Telecommunications, specializing into biotechnology and networking research areas.
Luis de Pedro completed his M.Sc. and Ph.D. degrees in Telecommunications Engineering at Universidad Politécnica de Madrid, Spain. He is a part-time associate professor at Universidad Autónoma de Madrid, Spain, and his research interests are focused on the performance application analysis.
Ivan González received his M.Sc. degree in Computer Engineering in 2000 and his Ph.D. degree in Computer Engineering in 2006, both from Universidad Autonoma de Madrid, Spain. His research interests are focused on HPC applications.
Javier Tamames received his Ph.D. degree in Chemical Science and is a senior scientist at the National Center of Biotechnology, CSIC, Madrid, Spain.
Author Contributions
Conceived and designed the experiments: GR, JT. Analyzed the data: GR. Wrote the first draft of the manuscript: GR. Contributed to the writing of the manuscript: LdP, IG. Agree with manuscript results and conclusions: LdP, IG, JT, GR. Jointly developed the structure and arguments for the paper: LdP, JT, GR. Made critical revisions and approved final version: LdP. All authors reviewed and approved of the final manuscript.