
Abstract
Whole genome sequencing of methicillin-resistant Staphylococcus aureus (MRSA) strain isolated from Sudan has led to a great deal of information, which allows the identification and characterization of some pivotal proteins. The objective of this study was to investigate the penicillin-binding proteins, PBP and PBP2a, of SO-1977 strain to have insights about their physicochemical properties and to assess and describe the interaction of some phytochemicals against them in silico. PBP and PBP2a from MRSA’s Sudan strain were found to be of great resemblance with some other strains. G246E single-nucleotide polymorphism was reported and identified in the allosteric binding site positioned in the non-penicillin-binding domain. The docked compounds demonstrated good binding energies and hydrogen bond interactions with residue Ser404 which plays crucial roles in β-lactam activity. This finding would contribute significantly to designing effective β-lactam drugs, to combat and treat β-lactam–resistant bacteria in the future.
Introduction
Life-threatening infection of methicillin-resistant Staphylococcus aureus (MRSA) has become a global clinical problem and remains a major cause of morbidity and mortality among bacterial diseases. 1 Staphylococcus aureus have developed resistance against some β-lactams by expressing drug-insensitive penicillin-binding proteins (PBPs) and through the action of β-lactamase which is hydrolyzing β-lactam drugs preventing them from acting on their target. 2 Penicillin-binding proteins play a paramount role in bacterial cell cycle, by catalyzing the transpeptidation reaction in cell wall construction. In addition to the 4 types of PBPs (PBP1, PBP2, PBP3, and PBP4), MRSA has the potential to express PBP2a, which is resistant to the action of methicillin and some other β-lactams. 3 Among the structural domains, the transpeptidase (TPase) domain plays a crucial role in bacterial life.4,5 PBP2a is a product of the resistance gene mecA, which functions as a surrogate transpeptidase when other PBPs are inhibited. 6 Therefore, an urgent development of potential therapy is highly needed to overcome the multiple drug resistance that is experienced by most of the currently used antibiotics. In the present research, we have set our goals toward studying the possibilities of finding potential PBP2a inhibitors and to predicting the 3-dimensional (3D) structure of PBP2a as well as using many bioinformatics tools. Moreover, to discover potential inhibitors for the PBP2a, some phytochemicals were in silico screened using molecular docking; results revealed promising chemical interties with profound molecular interactions. This study provides deep insights and findings that would contribute to understanding the function of these proteins and developing novel therapeutics. Besides, this is the first in silico study of PBP and PBP2a proteins in MRSA SO-1977 isolated from Sudan.
Materials and Methods
Sample preparation and next-generation sequencing
Bacterial sample was collected from wound swab specimen from a patient in Soba Hospital, Khartoum (Sudan). The colonies were identified and MRSA strain was determined. Bacterial DNA was extracted using Qiagen DNA extraction kit (Qiagen, Hilden, Germany). Resultant DNA was photometrically determined using a NanoDrop spectrophotometer (Thermo Fisher, Waltham, MA, USA). DNA library was prepared by Nextera XT Kit (Illumina, San Diego, CA, USA), followed by amplification with 12 cycles of polymerase chain reaction. DNA libraries were quantified using Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Thereafter, DNA sequencing was performed using Illumina MiSeq platform (Illumina). 7
Bacterial genome assembly and annotation
Poor-quality and adaptor-containing reads were filtered and trimmed using BBTools version 36. 8 Good-quality sequencing reads were assembled using SPAdes version 3.5.0. 9 The protein-coding genes were first predicted using Prodigal 2.60, whereas their function was predicted using BLASTN 2.2.25+ and HMMER 3.0, for searching for various sequence or domain databases.10–12 The whole genome sequencing was used for annotation, using RAST (Rapid Annotation using Subsystem Technology) and NCBI (National Center for Biotechnology Information) Prokaryotic Genome Annotation Pipeline.13,14 The annotated genes were extracted from the RAST server into an excel table and manually compared for genomic features, 13 and then PBP and PBP2a proteins were extracted from the list of annotated genes and prepared to be ready for further analysis. The genomic data from this study were deposited publicly in DDBJ (DNA Data Bank of Japan)/ENA (European Nucleotide Archive)/GenBank databases under Accession: NFZY00000000, BioProject database: PRJNA385553, and BioSample database: SAMN 06894057.
Protein sequence extraction
The sequences of PBP and PBP2a of other strains of S aureus were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/). Furthermore, searches for similar protein sequences were conducted by BLAST with default search parameters (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Multiple sequence alignments and polygenesis
Alignment of PBP and PBP2a target proteins (SO-1977) was done separately for each protein against selected sequences from the most similar strains (of BLAST results). Alignments were done using Clustal Omega program https://www.ebi.ac.uk/Tools/msa/clustalo/ and results were shaded by BoxShade tool. (https://www.ebi.ac.uk/services, http://www.ch.ebnet.org/software/BOX_form.html). After that, phylogenetic trees were constructed using MEGA6 software version 6. 15
Nonsynonymous SNP analysis
To analyze the functional effects of nonsynonymous single-nucleotide polymorphisms (nsSNPs) in PBP and PBP2a, 3 algorithms were used: SIFT (Sorting Intolerant From Tolerant; http://sift.jcvi.org/), PROVEAN (Protein Variation Effect Analyzer; http://provean.jcvi.org), and PANTHER (Protein Analysis Through Evolutionary Relationships; http://www.pantherdb.org/). These tools predict whether an amino acid substitution has an impact on the biological function of a protein grounded on the alignment-based score. The SIFT scores range from 0 to 1; scores ⩽0.05 are predicted, by the algorithm, to be damaging amino acid substitutions, whereas scores >0.05 are considered to be tolerated. Furthermore, if the PROVEAN score ⩽−2.5, the protein variant is predicted to have a deleterious effect, whereas if the PROVEAN score is >−2.5, the variant is predicted to have a “neutral” effect. In addition, the PANTHER score is interpreted as follows: “probably damaging” (time >450 myr, corresponding to a false-positive rate of ~0.2 as tested on HumVar), “possibly damaging” (450 myr > time > 200 myr, corresponding to a false-positive rate of ~0.4), and “probably benign” (time <200 myr). 16 Moreover, the functional (damaging) amino acid substitutions were resubmitted to HOPE server to get more information about damaging variants and their role in proteins’ structure and function (http://www.cmbi.ru.nl/hope/method).
Primary structure (sequence analysis)
Target protein sequences (PBP and PBP2a) were submitted to the ProtParam server (http://web.expasy.org/protparam/) to determine the molecular weight (MW), isoelectric point (pI), amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index (AI), and grand average of hydropathicity (GRAVY). 17 Percentages of hydrophobic and hydrophilic residues were manually calculated from the percentage of the amino acid composition. Target protein sequences were resubmitted to the SOSUI server (http://harrier.nagahama-i-bio.ac.jp/sosui/sosui_submit.html) to predict the transmembrane domains. 18 In addition, the Pfam server (http://pfam.xfam.org/) was used to predict all functional sites (domains) located within target proteins. 19
Secondary structure prediction
The secondary structure of PBP and PBP2a proteins was estimated by the SOPMA (Self-Optimized Prediction Method with Alignment 20 ; https://npsa-prabi.ibcp.fr/NPSA/npsa_sopma.html).
Homology modeling and model validation
The 3D structures of the PBP and PBP2a were constructed using Swiss-Model server (https://swissmodel.expasy.org/). The model’s quality was assessed using the QMEAN, ANOLEA, and GROMOS methods. 21 ERRAT server (http://servicesn.mbi.ucla.edu/ERRAT/) and Verify-3D (http://servicesn.mbi.ucla.edu/Verify3D/) were used to test the correctness of 3D protein models. 22 A residue-by-residue superposition was conducted for PBP2a, to check the alignment pattern of the 2 proteins using Molecular Operating Environment (MOE) package. A cutoff value of root-mean-square deviation (RMSD) is equal to 2.0 Å. 23
Molecular docking of PBP2a protein from SO-1977 strain
To investigate the possibility of finding potential inhibitors of the PBP2a, we have conducted virtual screening for various natural compounds from diverse classes using molecular docking. The compounds used in this study were extracted from the literature, 24 and they cover the following classes: lignans, coumarins, isoflavonoids, and chalcones. These classes of compounds, besides many phytochemicals, are known for their antibacterial activity. The docking study was conducted with the docking module of the MOE software. The compounds, 3D structures, were built with Chem3D software, then they were transferred and saved into MOE database, and the energy of each compound was minimized by an MMFF94x force field. Protein 3D structures were protonated and energy minimized using an MMFF94x force field. Finally, the 3D structure of the prepared protein was saved as a PDB file. A Gaussian Contact surface was drawn around the binding site. The receptor was verified as receptor and the site as ligand atoms. The placement method used was Triangle Matcher. The first scoring function was set to London dG and the refinement to force field. The docking process was then started by retaining 100 poses. The final refined poses were ranked by the MM/GBVI-binding free energy estimation. The depiction of results was generated by PyMol APBS Tools.
Results and Discussion
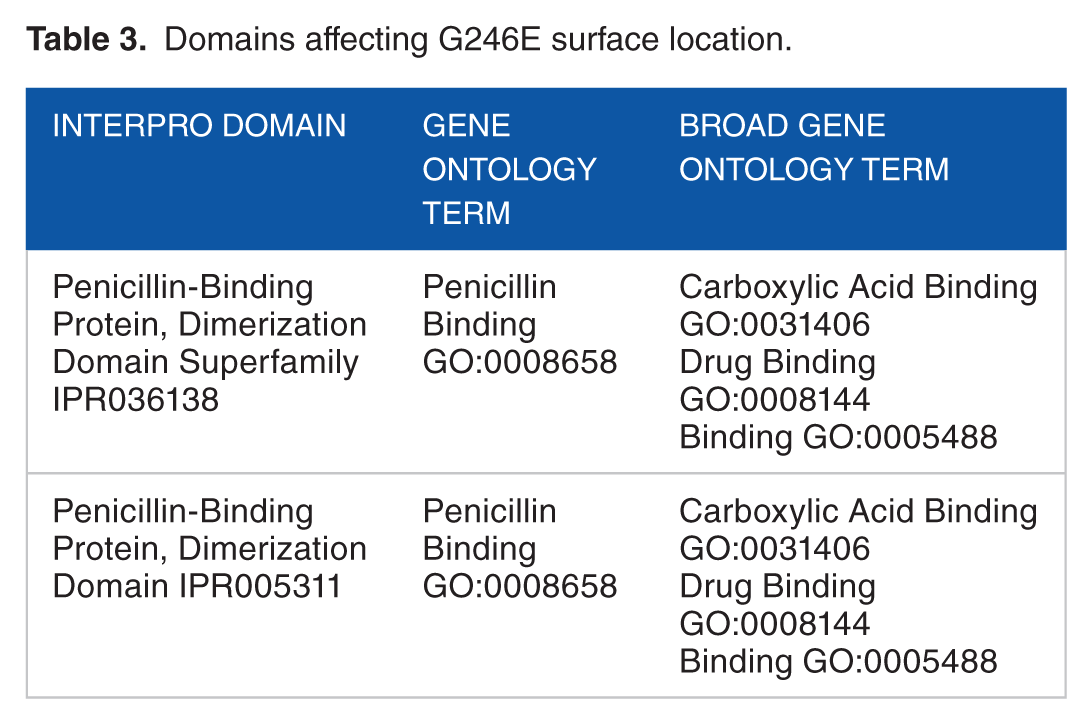
The BLAST alignment of PBP and PBP2a revealed a similarity range of 99% to 100% with the template proteins (Table 1). Alignment results showed that small numbers of amino acid substitutions were found to be present in PBP and PBP2a proteins of target strain (SO-1977). Within PBP protein, the P332S substitution was present only in the SO-1977 strain, whereas D480E, D662N, and S664T were present in SO-1977 strain and Staphylococcus aureus subsp. aureus 112808A (accession number: EOR49723). In PBP2a protein, only one substitution (G246E) was found in both SO-1977 and S aureus subsp. aureus (accession number: CCP89219) strains (Figure 1). Furthermore, all previous substitutions in PBP protein were found as a neutral and have no impact in resistant, but they might be of evolutionary effect. In PBP2a, the G246E substitution was predicted as functional (deleterious) as shown in Table 2. In this study, the G246E mutation was predicted in both SO-1977 and S aureus subsp. aureus, as it has previously been investigated in different MRSA strains by many researchers.25–28 The G246E mutation was located in non-penicillin-binding domain on allosteric binding site, as it might disrupt the interaction with other molecules. In accordance with the present results, Acebrón et al 29 reported and demonstrated that binding of β-lactams at PBP2a allosteric binding site, residing in the non-penicillin-binding domain, might play a paramount role in inhibiting its function, as it affects amino acid “residues” conformation in the main binding site; furthermore, mutations at this site have great influence in conferring resistance. According to the damaging substitution (Glycine into a Glutamine), the predicted result shows that each amino acid has its own specific size, charge, and hydrophobicity value in which the new amino acid is bigger, negative in charge, and less in hydrophobicity, compared with the wild type, which is smaller, neutral, and more hydrophobic than the mutant residue. The mutation introduces a charge at this position that it is suggested to cause repulsion between the mutant and neighboring residues. Moreover, as the residue is located on the surface of the protein, such mutation may disturb interaction with other molecules or even with other parts of the protein. In another side, the torsion angles for this residue are unusual; therefore, only Glycine is flexible enough to make these torsion angles and mutation in another residue will force the local backbone into incorrect conformation that would disturb the local structure (Figure 2). Furthermore, many motifs (ID numbers: DEG_COP1_1, DOC_WW_Pin1_4, TRG_ER_diArg_1) are annotated at this position where this mutation could cause damage (http://elm.eu.org/). This finding is in agreement with the results from previous studies focusing on β-lactam resistance, where the amino acid alterations, G246E, have been associated with an increase in resistance toward different β-lactams25–27,30,31 (Table 3).
Selected sequences of BLAST results.

Abbreviations: BLAST, Basic Local Alignment Search Tool; PBP, penicillin-binding protein.
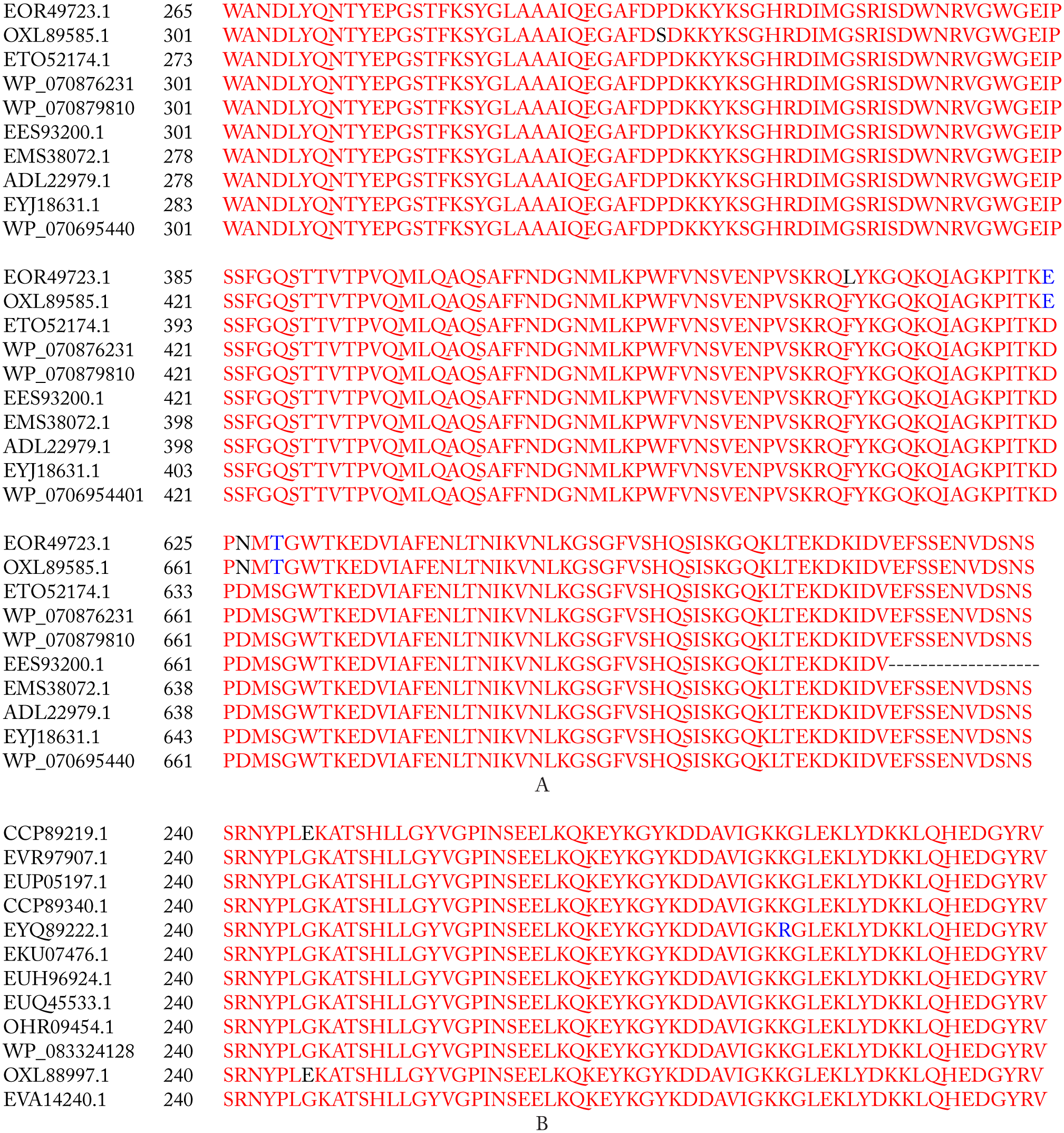
(A) MSA of the 9 PBP selected protein sequences, where OXL89585.1 is our target sequence. (B) MSA of the 11 PBP2a selected protein sequences where OXL88997.1 represents our target protein sequence. Red regions indicate similar residues, blue and black regions show amino acid substitution, and dashed regions indicate gaps. MSA indicates multiple sequence alignment; PBP, penicillin-binding protein.
Amino acid substitution affecting prediction

Abbreviations: PANTHER, Protein Analysis Through Evolutionary Relationships; PROVEAN, Protein Variation Effect Analyzer; PBP, penicillin-binding protein; SIFT, Sorting Intolerant From Tolerant; SNPs, single-nucleotide polymorphisms.

Close-up of the mutation (G246E). The protein is colored gray, and the side chain of both wild-type (in green color) and mutant (in red color) residues shows their target positions.
Domains affecting G246E surface location.
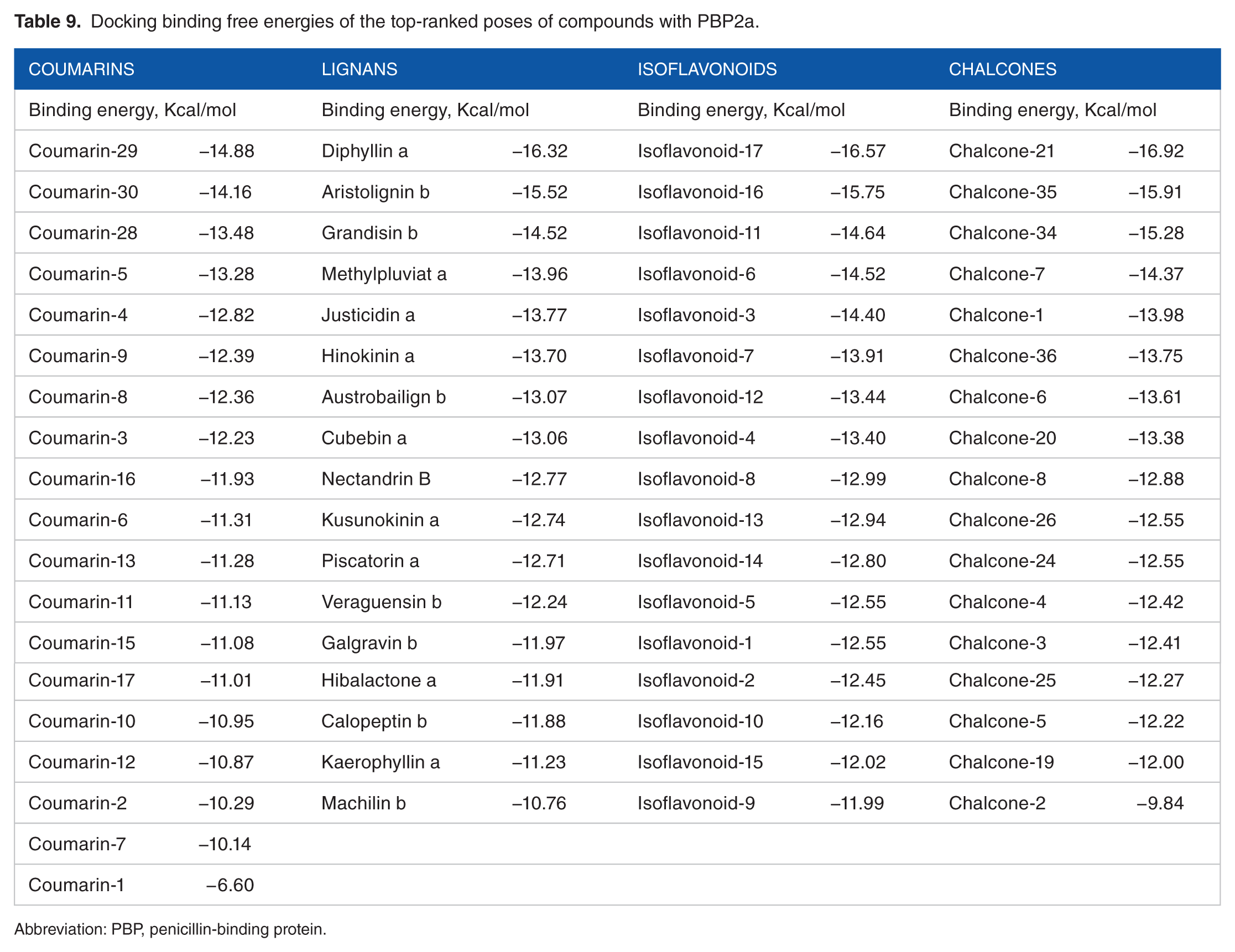
To depict the relationship among the retrieved protein sequences, phylogenetic trees were constructed. As can be seen from Figure 3, PBP sequence with accession number EOR49723 and PBP2a sequence with accession number CCP8219 were the closest sequences to the target PBP and PBP2a sequences, respectively. In respect to proteins’ amino acid composition, Leucine was the most abundant amino acid revealed which accounts for 12% to 13% of the protein’s primary structure and the least common amino acids were Tryptophan and Histidine which account for 1% with an absence of Cysteine amino acid (Table 4). According to the computed physicochemical characteristics of PBP and PBP2a proteins, the pIs of each were 9.28 and 8.62, respectively, which indicates that the proteins are likely to precipitate within basic buffers and maintained within a neutral buffer. Furthermore, negative GRAVY values of target proteins refer to hydrophilic nature (water-attracted). Instability indices for the 2 target proteins were below 40, which indicates that they would remain stable within a solution. In addition, the AI was quite high, which indicates that the proteins would remain stable over an array of temperatures (Table 5). Three different types of domains were found in each of PBP and PBP2a proteins, of which 2 (PBP dimer and transpeptidase) were shared between these target proteins, whereas PASTA and MecA domains were exclusively found in PBP and in PBP2a, respectively (Figure 4). Furthermore, the transmembrane regions of all proteins are rich in hydrophobic amino acids as can be seen from data in Table 6. Prediction of the secondary structures for the 2 proteins clearly showed that there were no disulfide bridges in the query sequence/structure (Table 7). The PBP modeling is based on the template PDB ID: 5tro, sequence identity was 99.47%, and the PBP oligo state is a homodimer. The PBP2a modeling is based on the template PDB ID: 1 MW, sequence identity was 100%, and the PBP2a oligo state is a monomer (Figure 5). Model validation was done by many tools, and the results were as follows: QMEAN and Z scores were −0.4 and 0.75 for PBP, whereas −0.69 and 0.68 for PBP2a. Furthermore, the quality of the PBP model was found to be 93% and 93.48% by ERRAT and Verify-3D, respectively, whereas that of PBP2a was 95.0% and 96.00% (Figure 6). The modeled PBP2a enzyme from SO-1977 strain has a nitrocefin molecule attached to its binding site, which was added during the homology modeling process. To evaluate the docking procedure and to inspect the protein-binding site for the pivotal residues, nitrocefin was re-docked into the binding site of PBP2a. Figure 8 presents the binding site of the protein model and selected residues within a 5-Å sphere and shows the crucial amino acid residues that surround nitrocefin molecule. It has been observed that the Ser404 residue has played a crucial role in the mechanism of the PBP2a interaction, as it is interacting covalently with nitrocefin molecule as depicted in Figure 8. The conformation of the docked poses of nitrocefin was compared with the initial conformation of the native ligand; the RMSD value was found to be 1.7 Å, which was less than the cutoff value (2.0 Å); and the docking binding energy of nitrocefin to the target enzyme was found to be −20.43 kcal/mol. The docking procedure was applied for 6 common β-lactam antibiotics, namely, penicillin G, amoxicillin, cephalosporin C, cefoxitin, thienamycin, and aztreonam. The binding energies of the enzyme-compound complexes resulted from docking are presented in Table 8. The scoring function binding was used as a basis for ranking the docked drugs in which all docked β-lactam antibiotics have revealed a similar binding profile. Among them, we have chosen amoxicillin to illustrate the pivotal binding interactions with PBP2a, as it has revealed a relatively higher binding energy value (−16.70 kcal/mol) compared with the other drugs; it was bound to the active site of the enzyme through a network of hydrogen bonding with the polar residues Ser404, Thr601, Ser599, His584, and Ser462 (Figure 9). Interestingly, the key amino acid Ser404, which plays the paramount role in the mechanism of β-lactam action, was found to be attached to the carboxylic hydroxyl of the amoxicillin molecule via a strong hydrogen bond at a distance of 1.3 Å. A total of 70 compounds belonging to lignans, coumarins, isoflavonoids, and chalcones were docked to the penicillin-binding domain of the PBP2a (Table 9); these ligands found in the binding site were used for directing the molecules to the binding site during the docking process. Docking scoring revealed that all the top-ranked compounds were docked at the binding site and interacted similarly, resembling the interaction pattern of amoxicillin with PBP2a. Chalcone-21, isoflavonoid-17, and diphyllin have demonstrated similar binding energy values of −16.92, −16.57, and −16.32 kcal/mol, respectively, whereas coumarin-29 revealed a relatively lower value (−14.88 kcal/mol) (Figure 10). Binding interactions of the top-ranked compounds are depicted in Figures 11 to 14; interaction with Ser404 was found to be the most dominant one. Chalcone-21 interacts with PBP2a residues through 3 hydrogen bonds. Figure 11B shows that the hydroxyl group of benzene ring forms hydrogen bond with Ser404 (1.86 Å), the hydroxyl group of the ring forms hydrogen bond with Ser401 (3.01 Å), and the α-, β-unsaturated carbonyl moiety forms hydrogen bond with Gln522 (2.85 Å). Isoflavonoid-17 has shown a total of 5 hydrogen bonds with the protein residues, the hydroxyl of the phenyl ring forms 2 hydrogen bonds with Ser404 (2.46 Å) and Thr601 (2.92 Å), the other hydroxyl of the same ring forms hydrogen bond with Thr601 (1.65 Å), and the hydroxyl of the other ring forms 2 hydrogen bonds with Glu448 (1.56 Å) and Ser462 (3.33 Å) (Figure 12B). Diphyllin interacts with PBP2a via 6 hydrogen bonds with 4 amino acid residues (Figure 13B). The hydroxyl group of the compound forms 2 hydrogen bonds with Ser404 (3.32 Å) and Ser463 (2.56 Å), and the 2 adjacent methoxy groups form 4 hydrogen bonds with Gln522 (2.31 and 3.17 Å) and Asn465 (3.1 and 2.53 Å). Finally, coumarin-29 has revealed 3 hydrogen bonds: one of the hydroxyl groups forms hydrogen bond with Ser404 (2.77 Å) and the other hydroxyl forms hydrogen bonds with both Asn465 (2.6 Å) and Gln522 (2.61 Å) (Figure 14D). The studied compounds are somehow of similar scaffolds, which reflects the resemblance in their binding energies and binding interactions. These scaffolds would be promising lead candidates acting as PBP2a inhibitors that might be subjected to further structural optimization to enhance their biological activity and hence been synthesized and tested to evaluate their actual biological activity. The current docking study was performed in the PBP2a-binding site, where β-lactams bind, in the absence of the resistance, and act via acylation of Ser404 (or Ser403 in other strains).30,32,33 However, when resistance occurs, β-lactam antibiotics undergo ring cleavage by the action of β-lactamase. 34 In this study, we have provided an evidence for the possibility of molecules other than β-lactams to be interacting with PBP2a enzyme through a network of H-bonding among them (interaction with the key residue Ser404). Although the G246E mutation plays pronounced role in the protein’s mechanism, however, using molecular docking is not possible to predict its vital role as there is no scoring function, and search algorithms can probe the remote action of this type of inhibition.
Amino acid composition.

Abbreviation: PBP, penicillin-binding protein.
Physicochemical parameters of PBP and PBP2a.

Abbreviation: PBP, penicillin-binding protein.
Transmembrane sequence analysis.

Abbreviation: PBP, penicillin-binding protein.
Secondary structure prediction of proteins.

Abbreviation: PBP, penicillin-binding protein.
Docking binding free energies of the top-ranked poses of β-lactams with MRSA PBP2a.

Abbreviation: PBP, penicillin-binding protein.
Docking binding free energies of the top-ranked poses of compounds with PBP2a.
Abbreviation: PBP, penicillin-binding protein.

(A) Phylogenetic tree of PBPs of different strains of Staphylococcus aureus. (B) Phylogenetic tree of PBP2a of different strains of S aureus. Branch in barn color mark protein sequences of target strain (SO-1977). PBP indicates penicillin-binding protein.

Schematic illustration showing the domains of PBP and PBPs of the target strain and their location. PBP indicates penicillin-binding protein.
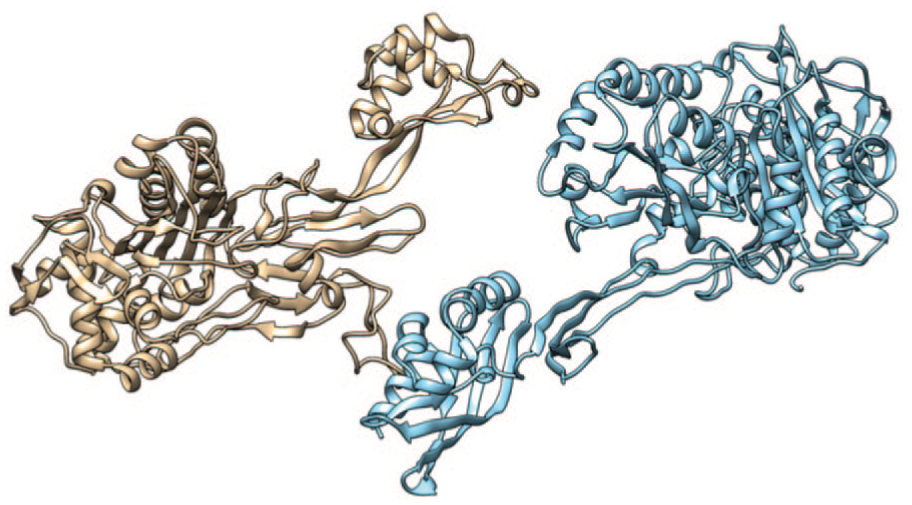
Three-dimensional structures of target PBPs using the Swiss-Model server. The model in brown color represents the PBP, whereas the blue one represents the PBP2a. Models explored and pictures exported were done using chimera. PBP indicates penicillin-binding protein.

Evaluation of PBP models by ERRAT server. “A” shows the quality model of PBP and “B” shows the quality model of PBP2a. PBP indicates penicillin-binding protein.

Cartoon representation of the 3-dimensional superposition of PBP2a of MRSA strain SO-1977 (shown as yellow) and template protein Staphylococcus aureus strain 27r PBP2a (shown as purple). The calculated RMSD value obtained from alignment was found to be 0.35 Å. PBP indicates penicillin-binding protein; MRSA, methicillin-resistant Staphylococcus aureus strain; RMSD, root-mean-square deviation.

Three-dimensional representation of the binding site of the modeled PBP2a strain SO-1977 complexed with nitrocefin: nitrocefin (carbon in pink) and amino acid residues (pale blue). PBP indicates penicillin-binding protein.

Docking of amoxicillin into Staphylococcus aureus strain SO-1977 PBP2a; protein residues are shown as sticks and colored by atom types, carbons are light blue, oxygens are red, and nitrogens are deep blue. Amoxicillin is shown as stick and its carbons shown as pink, oxygens as red, nitrogens as deep blue, and sulfur as yellow. PBP indicates penicillin-binding protein.

Chemical structure of the top molecules.
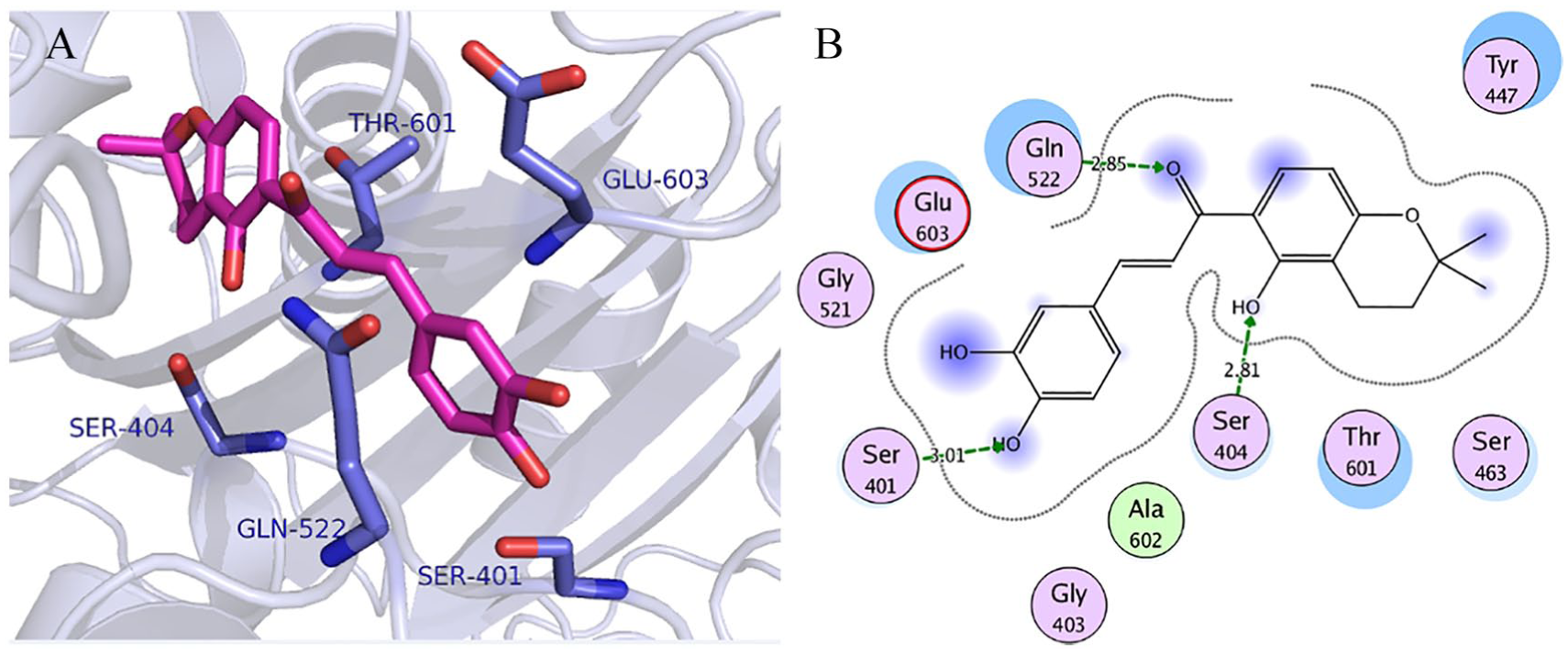
Visualization of binding interactions of chalcone-21 in the binding site of resistant SO-1977-PBP2a model: (A) 3-dimensional representation and (B) 2-dimensional representation. PBP indicates penicillin-binding protein.
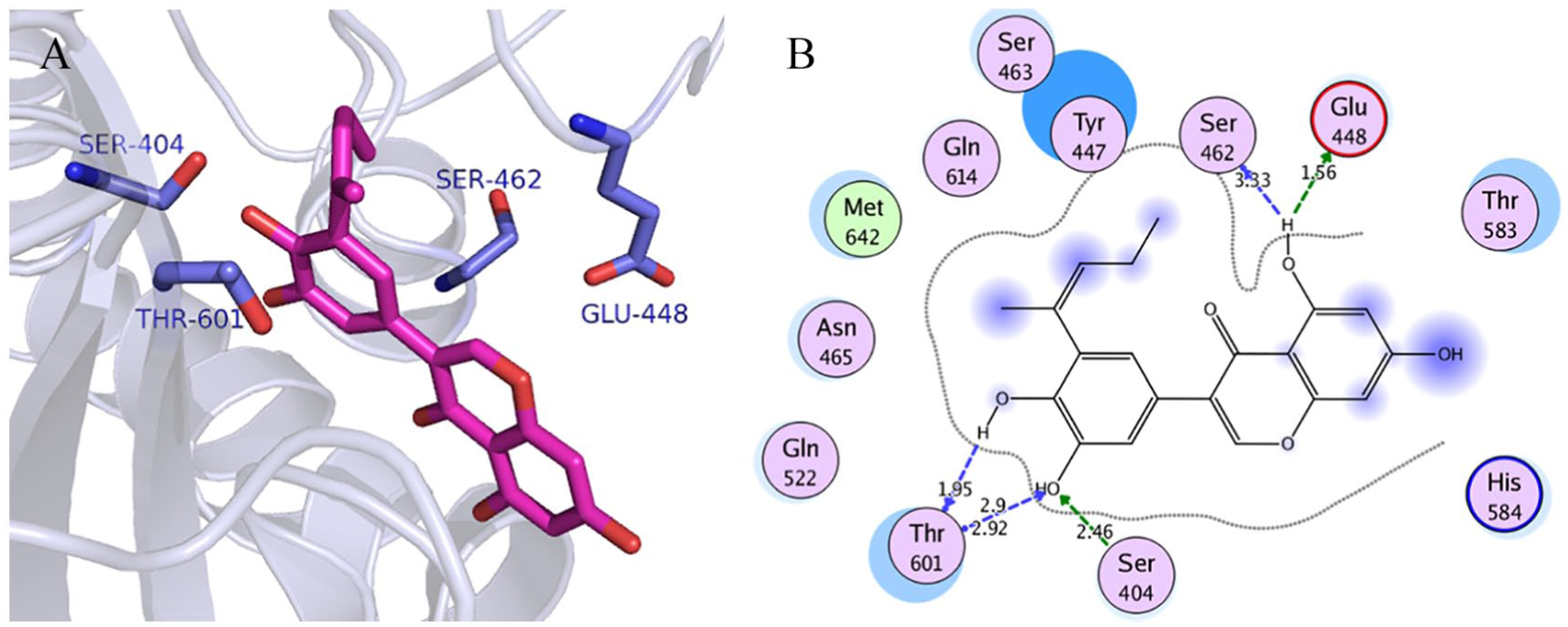
Visualization of binding interactions of isoflavonoid-17 in the binding site of resistant SO-1977-PBP2a model: (A) 3-dimensional representation and (B) 2-dimensional representation. PBP indicates penicillin-binding protein.

Visualization of binding interactions of diphyllin in the binding site of resistant SO-1977-PBP2a model: (A) 3-dimensional representation and (B) 2-dimensional representation. PBP indicates penicillin-binding protein.

Visualization of binding interactions of coumarin-29 in the binding site of resistant SO-1977-PBP2a model: (A) 3-dimensional representation and (B) 2-dimensional representation. PBP indicates penicillin-binding protein.
Conclusions
Using the next-generation sequencing and bioinformatics approaches is nowadays being vitally important and powerful tool in diagnosis of diseases; therefore, it provides a pronounced opportunity for further research to be performed to discover new therapeutics against challenging diseases. This is the first study in Sudan and may constitute a possible candidate for further genetic studies. This work could facilitate the detection and identification of the challenges related to the antibiotics resistance and helps in the development of new antimicrobial drugs.
Footnotes
Acknowledgements
The authors are grateful to Professor Qurashi M. Ali, the National University (Sudan) president, for his encouragement and support and also thankful to Ms Sumaya Kambal for review and editing the final draft of the manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out with the support from the National University Sudan.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SBM planned and directed the project, prepped the sample, and did the next-generation sequencing. SBM, MAB, TAA, NIA, M-ABE, NAK did the in silico analysis. SBM, MAB, ZSSA, and TAA wrote the first draft of the manuscript. SBM, MAB, and TAA contributed to the writing of the manuscript, agreed with manuscript results and conclusions, and made critical revisions and approved final version. All authors reviewed and approved the final manuscript.
Ethical approval and consent to participate
This research was approved by the Ethical Committee of the International University of Africa, Khartoum, Sudan, and written consent was obtained.
Availability of Data and Materials
The data sets used and/or analyzed during this study are available from the corresponding author on reasonable request.