
Abstract
Plant disease resistance genes (R-genes) play a critical role in the defense response to pathogens. Barley is one of the most important cereal crops, having a genome recently made available, for which the diversity and evolution of R-genes are not well understood. The main objectives of this research were to conduct a genome-wide identification of barley Coiled-coil, Nucleotide-binding site, Leucine-rich repeat (CNL) genes and elucidate their evolutionary history. We employed a Hidden Markov Model using 52 Arabidopsis thaliana CNL reference sequences and analyzed for phylogenetic relationships, structural variation, and gene clustering. We identified 175 barley CNL genes nested into three clades, showing (a) evidence of an expansion of the CNL-C clade, primarily due to tandem duplications; (b) very few members of clade CNL-A and CNL-B; and (c) a complete absence of clade CNL-D. Our results also showed that several of the previously identified mildew locus A (MLA) genes may be allelic variants of two barley CNL genes, MLOC_66581 and MLOC_10425, which respond to powdery mildew. Approximately 23% of the barley CNL genes formed 15 gene clusters located in the extra-pericentromeric regions on six of the seven chromosomes; more than half of the clustered genes were located on chromosomes 1H and 7H. Higher average numbers of exons and multiple splice variants in barley relative to those in Arabidopsis and rice may have contributed to a diversification of the CNL-C members. These results will help us understand the evolution of R-genes with potential implications for developing durable resistance in barley cultivars.
Introduction
Plants have evolved complex signaling pathways for pathogen detection and defense response. 1 Lacking an adaptive immunity and cell-transporting circulatory system, plant resistance to pathogens depends upon innate immunity that utilizes molecular signaling to initiate local and systemic responses. 2 Resistance genes (R-genes) encode proteins that detect pathogens.3,4 Plant immunity can be divided into two types: pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and effector-triggered immunity (ETI).2,5 PAMPs are pathogen structural molecules, such as bacterial flagellin, peptidoglycan, and fungal chitin, that the plant's immune system perceives through membrane-localized, receptor-like kinases called pattern recognition receptors, which elicit a response.6,7 In contrast, ETI involves the interaction between specific pathogen effectors and NBS-LRR receptors within the cell. 5 Resistance responses vary widely and act in limiting the spread and effectiveness of the pathogen 3 including the following: (1) causing localized death of infected tissue through hypersensitive response, 8 (2) promoting hostile conditions for pathogens such as hydrogen peroxide production in an oxidative burst, 9 and (3) fortifying cell walls to strengthen the physical barrier between pathogens and the plant protoplasm. 10 Resistance responses are expensive for the cell 11 ; therefore, in the absence of a pathogen, diverse control factors are mobilized, 12 including salicylic acid production for localized and systemic resistance,13,14 WRKY transcription factors, 15 and silencing through micro-RNA. 16
Several models have been proposed to describe the mechanism of host–pathogen relationships. The gene-for-gene model involves direct interaction between a single pathogen avirulence gene and a plant R-gene. 17 Additionally, there is evidence of indirect interaction as described in the guard model, where R-proteins bind with or guard particular target proteins, activating a response when the guarded protein is cleaved or modified by a pathogen.18,19 Similar to the guard model, the decoy model describes specific decoy proteins that mimic unguarded pathogen effector targets, forming a complex with effectors that is perceived by NBS-LRR R-proteins. 20 With increasing understanding of molecular interactions between the pathogen and host, the zig-zag model was proposed to describe coevolution of plant R-genes and pathogen effectors. 2 In this model, the pathogen evolves effectors to reduce the effectiveness of the plant's PTI response, and the plant responds to these newly evolved effectors by developing receptors that initiate ETI. 2 Intense selection pressures from pathogens cause R-genes to evolve rapidly through several mechanisms, including recombination and transposable ele-ments.4,21,22 However, R-genes can also be removed from the genome through loss of lineages and deficient duplications. 23 R-genes have been recently classified into eight major groups: (1) Toll interleukin receptor, Nucleotide-binding site, Leucine-rich repeat (TIR-NBS-LRR or TNL); (2) Coiled-coil, NBS, LRR (CC-NBS-LRR or CNL), (3) LRR transmembrane domain (LRR-TrD); (4) LRR-TrD-kinase; (5) LRR-TrD protein degradation domain proline-glycine-serine-threonine (LRR-TrD-PEST); (6) TrD-CC; (7) TNL-nuclear localization signal amino acid domain (TNL-NLS-WRKY); and (8) enzymatic genes. 24 Among these groups, the NBS-LRR (TNL and CNL) genes form the largest group and respond to various pests and pathogens. 25 The NBS-LRR genes are highly variable,25,26 but their NBS region contains several conserved motifs that can be traced back to early land plant groups. 27 The N-terminal region of the protein contains either a TIR or a CC region, the former being restricted to only dicot species. 26 The NBS contains a highly conserved Nucleotide-Binding domain shared by Apaf-1, resistance gene products, and CED-4 (NB-ARC), 28 whereas the C-terminal LRR is a highly variable region that can bind to many different molecules.7,29 The CNL genes have been identified in the genomes of many plant species: 52 in Arabidopsis, 26 159 in rice,30,31 188 in soybean,30,32 203 in grape, 33 65 in potato, 34 94 in common bean, 35 177 in alfalfa, 36 six in papaya, 37 and 18 in cucumber. 38 Recent studies have shown that CNL genes are effective at resistance to the devastating Ug99 stem rust strain in wheat.39,40 In the present study, we explored the recently available barley genome 41 to understand the diversity and evolution of CNL genes.
Cultivated barley (Hordeum vulgare L.) is a grass family (Poaceae) member that was domesticated approximately 10,000 years ago 42 and is now a major cereal crop. 43 Even before genomic information was available, the use of barley cultivars with R-gene Rpg1 in 1942 greatly reduced the loss of barley yield due to stem rust, Puccinia graminis, in the Midwestern United States and Canada.44,45 Additionally, barley cultivars containing the gene Rph20 are resistant to barley leaf rust (pathogen: Puccinia hordei), which otherwise causes up to 62% crop loss.46,47 It has been shown that the recessive barley mlo mutant allele confers broad-spectrum resistance to powdery mildew (pathogen: Erysiphe graminis f. sp. hordei),48,49 but the presence of these mlo mutant alleles also increases susceptibility to Ramularia Leaf Spot (RLS). 22 Genes within the mildew locus A (MLA), some of which are CNL, also play a role in resistance to powdery mildew and were formed through duplication, inversion, and insertion over a period of greater than seven million years. 50 It has been hypothesized that many variants of MLA are different alleles rather than separate genes. 51 In a recent study, higher nucleotide diversity was found in wild barley samples relative to that in the cultivated samples. 52
The objectives of this research project were to identify CNL R-genes in the barley genome and elucidate their evolutionary relationships. This in silico analysis aims at comparing barley CNL genes with their orthologs in rice and Arabidopsis thaliana. With barley and related species making up a significant portion of the staple food supply, analyses that would potentially lead to more pathogen-resistant cultivars make a significant contribution to agriculture. Wheat, another member of the same family, may contain many similar R-gene pathways and barley resistance may be conferrable to the wheat cultivars.
Materials and Methods
CNL gene identification
Barley CNL gene identification followed methods used in Arabidopsis 26 and soybean. 53 Barley protein sequences were accessed through the Ensembl Genomes database. 54 Arabidopsis CNL genes, as identified and classified by Meyers et al. 26 , were obtained from Phytozome, 55 and their orthologs in rice were obtained, as confirmed in the study by Benson. 31 Fifty-two Arabidopsis CNL genes were used as reference sequences to explore orthologs in the barley genome (62,236 analyzed protein sequences), by aligning the sequences in the program ClustalW 56 and constructing a Hidden Markov Model (HMM) using HMMER version 3.1b2 57 at a stringency of 0.05. Further selection involved identification of NB-ARCs using the database Pfam, 58 accessed through InterProScan. 59 Genes containing NB-ARCs were then aligned using ClustalW, integrated within the program Geneious. 60 A second HMM profile was constructed to use these barley NB-ARC-containing proteins to perform a reiterative search of the genome with a stringency of 0.001. InterProScan 59 was then used to identify the protein sequences with both an NB-ARC and a DiseaseResist region. MEME analysis, 61 set to display the 20 most prevalent motifs, was used to identify protein sequences with P-loop, Kinase-2, and GLPL regions, the diagnostic motifs of CNL genes.
Phylogenetic analysis
NB-ARCs were extracted from the protein sequences identified by the MEME search. These sequences were aligned using ClustalW integrated within the program Geneious. The protein sequences were imported along with the original Arabidopsis genes and their orthologs in rice for phylogenic comparison. An evolutionary model for the CNL amino acid sequences was determined using a maximum-likelihood model test function in the program MEGA 6.0, 62 which identified JTT+G+I as the best substitution model. This model was used to construct a maximum-likelihood tree with 100 bootstrap replicates.
Gene structural variation, clustering, and Ks analysis
Information on location and exon size was obtained from Ensembl Genomes, which was uploaded into the program Fancy gene v1.4 63 to generate an exon map. Entire chromosome sequences were accessed through Ensembl Genomes and imported into the program Geneious. A genomic map to visualize gene clustering was generated by matching gene locations with their respective chromosomes, along with centromere locations. 41 Nucleotide intervals between genes on each chromosome were determined in order to quantify any clustering following the study by Jupe et al. 64 Accessions were grouped into clades according to their nesting pattern. Coding sequences were downloaded from Ensembl Genomes to estimate the nonsynonymous substitutions per nonsynonymous site (Ka) and synonymous substitutions per synonymous site (Ks) values, and Ka/Ks ratios were calculated using the program DnaSP 5.10.1. 65 Average Ks values were used to infer the relative time of duplication events.
Results
Identification of CNL genes
CNL gene clusters in the barley genome: 15 clusters containing 39 genes were identified using a sliding window of 200 kb and eight open-reading frames (ORFS).

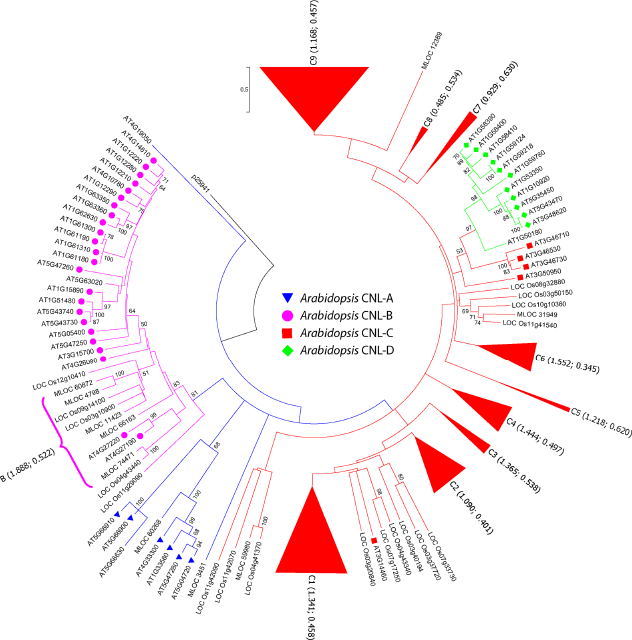
Phylogenetic analysis of the CNL genes from H. vulgare (MLOC), Arabidopsis (AT), and Oryza sativa (LOC). The maximum-likelihood tree was constructed using the JTT+G+I model with 100 bootstrap replicates. Arabidopsis CNL-A, CNL-B, CNL-C, and CNL-D groups are represented as blue triangles, pink circles, red squares, and green diamonds, respectively. The tree was rooted using outgroup p25941 as used in Arabidopsis. 26 CNL-C clades were collapsed to increase readability (for the complete tree, see supplementary Fig. 1), and the list of genes can be found in supplementary Table 1 The Ks values and Ka/Ks ratios are shown in parentheses following the clade name, first Ks and then Ka/Ks ratio. The collapsed clades contain only barley and rice genes with the exception of clades C2 and C6, containing Arabidopsis orthologs AT3G14470 and AT3G07040, respectively.

Maximum-likelihood phylogenetic analysis of MLA accessions and selected barley CNL-C9 gene members using the JTT+G+I model with 100 bootstrap replicates. The tree was rooted using outgroup p25941 as previously used in Arabidopsis. 26
Phylogenetic relationships
Phylogenetic relationships of barley CNL genes and their orthologs in Arabidopsis are shown in Figure 1 (also in Supplementary Fig. 1 and Supplementary Table 1). Among the four clades previously reported in dicot species,26,31 CNL-D is completely absent in barley. The vast majority of the barley CNL genes (168 of the 175 members) belong to the clade CNL-C. Very few members of the CNL-A (two members) and CNL-B clades (five members), as well as the large amount of the CNL-C genes in barley, were consistent with those in rice, but diverse from Arabidopsis (Fig. 1). The orthologs in rice and barley show a high degree of interspecific nesting with a diversified CNL-C clade and complete absence of CNL-D members. Basal support for CNL-C is weak but leaf branches with specific gene relationships are strongly supported (BS >90%). Identification of MLA genes using BLAST within the Ensembl Genomes database showed that MLOC_10425 and MLOC_66581 are the likely accession names for many MLA sequences (Fig. 2).
MEME analysis, gene clustering, and structural variation
CNL orthologs of barley, rice, and Arabidopsis with associated pathogens.
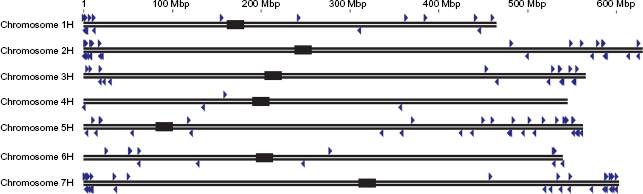
Distribution of the CNL genes on the chromosomes of barley (N = 7). The black lines and the blue arrows represent chromosomal length and gene location/orientation, respectively. Black rectangles indicate the centromere positions on each chromosome.
Gene locations on each chromosome were visualized to show CNL gene clustering (Fig. 3), which is defined as: (1) genes within a 200 kb sliding window and (2) fewer than eight other genes between the beginning and end of the cluster. Using these criteria, 15 gene clusters were identified (Table 1). Genes tended to be located in the extra-pericentromeric regions of chromosomes (Fig. 3). Each chromosome except chromosome 4H contained at least one cluster, and 10 of the 15 clusters were composed of only two genes, as shown in Table 1.
Ks values
Synonymous substitutions per synonymous site (Ks values) are often used as a proxy for inferring duplication events, so we used Ks values in inferring relative age of the CNL gene clusters (Table 1). Average Ks values were highest for CNL-B members and lowest for the CNL-C8 members (Fig. 1). All average Ka/Ks ratios were less than 1, indicating a prevalence of purifying selection. Functional homologs for the identified barley genes were compiled and compared with results from the phylogenetic analysis (Table 2). Using this information, instances of genomic expansions as well as reductions were inferred.
Discussion
Phylogenetic analysis and evidence of duplications
Phylogenetic analysis of the CNL protein sequences from barley and Arabidopsis showed a high level of tandem duplications within each species. Barley R-genes were nested as expected within the CNL-A, CNL-B, and CNL-C clades with their orthologs in Arabidopsis, concurring with the previous findings in rice 31 and Aegilops tauschii. 66 We observed fewer members of CNL-A and CNL-B, and complete absence of CNL-D in barley relative to that in Arabidopsis. Using comprehensive phylogeny of flowering plants 67 as a reference, we infer that Arabidopsis has experienced a reduction in CNL-C and expansions in CNL-A, CNL-B, and CNL-D. In a recent analysis of CNL genes in soybean (Glycine max), 32 a similar expansion in the CNL-C clade was observed. In contrast to CNL genes in soybean, we found a sharp reduction in CNL-A and CNL-B, and absence of CNL-D, in both barley and rice, which may be common in other grass species as well. Phylogenetic analysis of CNL genes of barley with rice (a model monocot 68 with a more recent common ancestor 69 ) showed more interspecific nesting patterns than with Arabidopsis (Fig. 1). Existing differences in R-gene diversity, structure, and evolutionary rates across these species may reflect phylogenetic constraints and species-specific evolutionary history. 70
Closely related genes within the same gene cluster in the phylogenetic tree (Fig. 1 and Table 1) show strong evidence of gene duplication events. Despite the huge genome size (5.1 Gb) of barley, there are numerous closely located CNL genes and their clusters that diversified through tandem duplications. One of the most striking examples of tandem duplication involves MLOC_24729 and MLOC_44743 genes, which are only 113 bases apart and are 69.5% identical (528 of 760 sites). The gene accessions MLOC_19475, MLOC_58383, MLOC_44175, and MLOC_12318 are closely related and form their own clade (Fig. 1), with three of these genes located within a 2.24 Mb segment of chromosome 7H, another instance of tandem duplication. The fourth gene in the same clade, MLOC_12318, is located on chromosome 2H, indicating that it resulted from segmental duplication. Similar duplication events have been reported in other plant genomes. 71 Overall variation within R-genes is attributed to duplications, recombination, and diversifying selec-tion, 25 with whole-genome duplications lessening selective pressures and allowing for diversification, as seen in the soybean genome. 72 Increased diversity of R-genes may provide barley with a selective advantage even though maintenance of R-genes during low pathogen exposure might prove very costly as suggested in literature. 73 While not residing within a technically defined cluster in barley, many genes are likely formed by gene duplication events, the origin of which could be traced to a common ancestor gene. The genes MLOC_11112, MLOC_30912, and MLOC_15443 form their own clade, with MLOC_30912 basal to the other two. MLOC_11112 and MLOC_30912 are clustered on chromosome 7H, likely formed by tandem duplication. The third gene, MLOC_15443, is approximately 1 Mb upstream of the other two, a possible instance of segmental duplication. Another example is a five-gene subclade (MLOC_66610, MLOC_66596, MLOC_19284, MLOC_68128, and MLOC_3117; BS 78%) in which all five genes are located within a 2.1 Mb section of chromosome 1H, likely to have arisen through gene duplication. It has been shown that R-genes can cluster in larger regions that do not fall within the defined criteria (ie, with the narrow sliding window) of a cluster. 74 In Medicago, superclusters have been identified in which a single-chromosome arm contains a large percentage of the genome's R-genes. 36 Zhou et al. 30 suggest that duplications of diversely clustered R-genes could explain the frequent and dissimilar duplications.
Ks values have been used to infer the history of duplication events within a genome, especially when analyzing genome duplications or polyploidy.75,76 The barley CNL-B clade has a higher average Ks value than any CNL-C subclade, suggesting recent expansion of CNL-C members in grasses (see Fig. 1). While average Ka/Ks values for each CNL-C clade were <1 indicating purifying selection, 23 individual pairwise values were >1, 15 of those being from CNL-C9. This indicates that while the majority of the identified genes are undergoing purifying selection, a few genes are undergoing positive selection. These Ks values can also give insight into the clustered genes that arise from duplications. For instance, cluster 3_1, composed of MLOC_56904 and MLOC_56905, has a very low Ks value of 0.217, indicating a recent duplication event. Since rice only has one paralog to these two sequences, LOC_Os01g05620 (Fig. 1), the duplication event likely happened after the split of rice and barley lineages. A similar case is shown by MLOC_44743 and MLOC_24729 (cluster 2_1), which have the Ks value of 0.194 and do not have a close paralog in rice, suggesting more recent evolution after rice and barley split. The same happens with cluster 7_3 (MLOC_6883 and MLOC_31061) with a low Ks value of 0.163. From this information, it can be inferred that cluster 3_1 formed first, followed by cluster 2_1, and finally 7_3.
Arabidopsis and rice homologs in barley
Looking more closely at the gene duplications and expansions within the barley genome, a species-specific history of pathogen load can be inferred. Arabidopsis gene AT3G07040 is functionally known as RPM1, an NBS-LRR gene that recognizes either the AvrRpm1 or AvrB type III effectors of Pseudomonas syringae, conferring resistance through a hypersensitive response. 77 As shown in Figure 1 and summarized in Table 2, barley contains 17 homologs (clade CNL-C6) of RPM1, what we infer to be a large expansion. It is possible that monocots faced a heavy P. syringae load during their evolutionary history, perhaps both before and after barley and rice diverged, since rice contains only 11 RPM1 homologs (Table 2). Another possibility is that Arabidopsis experienced a reduction through pseudogenization. In some other cases, the barley genome contains fewer R-genes than Arabidopsis. The Arabidopsis ADR1 genes (AT1G33560, AT4G33300, AT5G04720, and AT5G47280) are involved in the resistance response to Peronospora parasitica and Erysiphe cichoracearum. 78 The barley genome contains only one homolog (ie, MLOC_60268) for these four genes in Arabidopsis. The same occurs with RPP8 and RPP13 where many Arabidopsis gene members do not have any homologs in barley. Barley and rice appear to differ in the number of ZAR1, RPP13, and ADR1 homologs, with barley's single ADR1 homolog not being represented in the rice genome. There are also no barley homologs for AT1G10920 (LOV1 – CNL-D), which causes susceptibility to Cochliobolus victoriae. 79
The MLA genes in barley confer resistance to powdery mildew (Blumeria graminis f. sp. hordei). 80 We have identified many variants of MLA in our analysis (see Fig. 2). Two CNL-C9 gene members, MLOC_66581 and MLOC_10425, are highly similar to many different MLA sequences, with MLOC_66581 being a gene that most likely responds to powdery mildew. A BLAST search using MLOC_66581 and MLOC_10425 within the Ensembl Genomes database reveals that these two genes have the highest sequence identity to all MLA sequences. Seeholzer et al. 80 identified two functional MLA genes, MLA27 and MLA18, that both correspond to MLOC_66581 and MLOC_10425 accessions, respectively. As shown in Figure 2, these genes nested close to the MLA sequences, along with MLOC_64444 and MLOC_21734, which would also be closely related to the MLA genes. Thus, our results support the previous predictions by Shen et al. 51 and Seeholzer et al. 80 that many MLA variant sequences are alleles rather than separate genes.51,80
MLOC_60268 and MLOC_3451 are the only barley genes that nest with Arabidopsis CNL-A, with high bootstrap support. This shows that these two genes represent current CNL-A members in barley and are likely to have existed before the evolutionary split between monocot and dicot plants, between 200 and 140 million years ago.69,81 Accession MLOC_3451 shows most homology to the Apoptotic Protease-Activating Factor 1 (APAF1) from Triticum urartu, contributor of wheat's A-genome. 82 The similarity is not partial; entire protein sequence alignment shows that the sequences are 96.3% similar (1002 identical sites out of 1040). The presence of APAF1 would be expected since hypersensitive response involves an apoptosis-like cell death to prevent the spread of a pathogen. Therefore, CNL-A members in barley are predicted to contribute in hypersensitive response.
Gene structure and genomic content
Since there is no strict correlation between CNL gene content and genome size, a reasonable prediction of barley's CNL gene content could range from a few dozen members to a several hundred. Two earlier studies in barley reported 50 CNL genes 45 and 191 NBS-LRR genes. 41 While the rice and barley genomes have vastly different sizes, 420 Mb and 5.1 Gb, respectively,41,83 the genome-wide CNL diversity is rather similar, 159 and 175 genes, respectively. The P-loop, Kinase-2, and GLPL motifs are highly conserved in both species 30 and the RNBS A, B, and C motifs (Supplementary Fig. 2 and Supplementary Table 2) are also prevalent and conserved within the CNL genes.26,30
The CNL genes in barley showed a higher number of exons (3.34 exons per gene; Supplementary Fig. 3) than Arabidopsis and rice, with Arabidopsis genes generally consisting of one exon each 26 and rice averaging 2.1 exons per gene. 31 The higher number of exons per gene in barley could enable a more variable response to pathogens through multiple splice variation. Since many of the 982 initially identified protein sequences were variants of the same genes, it is possible that barley has used multiple splicing patterns to vary its pathogen-response proteins. It has been shown that NBS-LRR genes go through alternative splicing in Arabidopsis 84 and ratios of different transcripts are required for a resistance response. 85
While the number of exons per gene is higher than other species, the amount of CNL gene clustering is lower in barley, where only 39 of 175 CNL genes form 15 gene clusters (Fig. 3 and Table 1). In Arabidopsis, 109 of the 149 NBS-LRR genes formed 43 clusters, 26 but it was predicted that larger genomes may have a more complex distribution of CNL genes and that unclustered CNL genes are not unusual. 26 Barley genes that are highly clustered, such as those on chromosome 7H, allow for higher recombination rates and faster evolution.26,86 R-genes show varying speed of evolution, with Type I genes evolving relatively faster than Type II genes. 87 The expansion of CNL-C indicates that many of the CNL genes in barley are of the Type I class, suggesting a potential expansion in all grass species. Combining the evidence of duplications and clustering with Ka/Ks ratios, we see that the majority of barley CNL genes are currently undergoing purifying selection, which has been reported to be a common phenomenon among duplicated genes, especially in crop species. 88 The reduction in nucleotide diversity that took place during the cultivation of barley also likely impacted evolution of R-genes. 52
Current challenges in the development of durable resistance and future directions
Understanding of disease resistance has expanded greatly due to advances in molecular techniques and computational ability. Challenges regarding how efficiently we utilize genomic data to develop a more durable resistance continue to exist and can be overcome through the development and utilization of transcriptomic and metabolomics data. Additional genomic annotations are also needed as some chromosomal locations could not be accessed to determine clustering, and standardization of nomenclature is necessary. Specifically in the case of barley, current proteomic information is not complete and additional data would allow us to assess functionality. This, along with expression data upon pathogen exposure, and biochemical assays of signaling pathways are the major areas that require continued research. Also, cultivar-specific genome sequences would be useful to determine variation and educate breeders about how variation across cultivars is related to crop yield. This would allow for the development of barley cultivars that can better combat pathogens and may indirectly uncover directions for developing durable resistance in wheat and other closely related species.
Conclusions
In this study, we have presented our findings on the diversity and evolution of CNL genes in barley. The 175 identified barley R-genes show evidence of gene duplications as well as expansions and reductions of the NBS-LRR clades. The CNL gene diversity in barley is slightly higher than in rice and more than three times that in Arabidopsis. Many RPM1 homologs could be identified, indicating substantial exposure to pathogens such as P. syringae in barley's evolutionary history. Our results also indicated that several previously identified MLA sequences are the allelic variants of two CNL genes (MLOC_66581 and MLOC_10425). Many splice variants and multiple exons per gene may have allowed rapid diversification of R-genes in barley, especially the members of the CNL-C clade. As expected, several gene clusters were found, especially in the extra-pericentromeric regions of chromosomes, a location that experiences high rate of recombination needed for rapid gene diversification. Further research should aim to measure expression levels of these genes upon pathogen exposure and assess if some of these CNL genes could be used in developing cultivars with durable resistance.
Author Contributions
Conducted data mining, data analyses, and drafted the manuscript: EJA. Conceived, designed, and supervised the research project, as well as revised the manuscript: MPN. Contributed in data analysis, interpretation, and drafting of the manuscript: SA, SN, RNR, YY. All authors reviewed and approved of the final manuscript.
Supplementary Materials
Supplementary Figure 1
Maximum-likelihood phylogenetic tree with uncollapsed clades. See Figure 1 for detailed information including evolutionary model, coloring pattern, and outgroup.
Supplementary Figure 2
Motif structure of the 175 H. vulgare CNL genes based on MEME analysis. The CNL-A, -B, and -C clades are in blue, pink, and red, respectively. The six characteristic motifs P-loop, Kinase-2, GLPL, RNBS-B, RNBS-A, and RNBS-C are specifically named, and the following 14 motifs are named based upon their amino acid residues.
Supplementary Figure 3
Exon–intron variation across 175 CNL R-genes in barley. This illustration was generated using the program Fancygene 1.4 after input from Ensembl Genomes transcript information. Genes are presented by clade. Thick gray bars and dashed lines represent exons and introns, respectively. On the lower right corner is the summary information on the abundance of exons.
Supplementary Table 1
List of identified CNL genes and their corresponding clades.
Supplementary Table 2
Sequence information with the conserved motifs as identified by MEME analysis.
Footnotes
Acknowledgments
The authors thank BV Benson and Brian Moore for their assistance in data analysis.