
Abstract
Inferring the effective population size and the pattern of selection signatures is of interest both from an evolutionary perspective and to improve models for mapping of quantitative trait genes. We used DNA samples of 61 sires and 486 progeny of the Hanwoo, genotyped by the Illumina Bovine SNP50 BeadChip, to analyze the genetic structure. Our study showed a persistent decline in effective population size throughout the period considered, but suggested a marked decline at one distinctive time point (100th generation) and two sharp decline intervals (50th-25th generation and 25th-10th generation). This pattern can be explained by Hanwoo formation and the modern breeding program. Our results revealed 95 regions exhibiting the footprint of recent positive selection at a threshold level of 0.01. We found an overlap of the 11 core regions presenting top
Introduction
Linkage disequilibrium (LD) has recently become the focus of intense study because so many factors affect it and are affected by it. LD describes the nonrandom association of alleles at two or more loci. Information on the structure of LD at the population level is critical for interpreting and applying results of genome-wide association studies (GWAS) and genomic selection for the improvement of economically important traits.1,2 LD can help to unravel the genetic relationships among diverse breeds and the phylogenetic relationships between domestic animals and their wild ancestors. LD can be used to enable effective exploitation of the potentially significant historical events that occurred during domestication, breed formation, and ongoing selection (eg, bottleneck effects, genetic drift, and selective sweeps).
A wide variety of statistics have been proposed to measure the amount of LD, and these have different strengths, depending on the context. In the current literature, the two most popular measures of LD between pairs of biallelic markers are
Effective population size (
Detection of selective sweeps is useful because the effect of selection on the distribution of genetic variation can be difficult to distinguish from the pattern of genetic variation that arises after certain demographic events.6,7 The long-range haplotype (LRH) test examines the relationship between allele frequency and the extent of LD.
8
LRH measures the decay of identity, as a function of distance, of haplotypes that carry a specified “core” allele at one end. Alternative methods to detect selective sweeps include Tajima's
The Korean native cattle (Hanwoo) is known for its marbled fat, tenderness, juiciness, and characteristic flavor. Hanwoo originated from a hybrid of
Materials and Methods
DNA samples
SNP markers throughout the Hanwoo genome were genotyped. Samples were collected from 547 bulls born from spring through fall of 2006 in Seosan, South Korea. The bulls were 61 sires and 486 progeny. The size of the 61 sire families ranged from 2 to 18 bulls per sire, with an average of 8 bulls. Pedigree information was obtained from the Hanwoo Improvement Center of National Agricultural Cooperative Federation, Seosan. The mean kinship (coefficient of coancestry) is 0.0119, and the mean inbreeding coefficient is 0.4%, determined using pedigree information from up to four generations. For the purposes of this study, these values were assumed to be zero. Coancestry and inbreeding coefficients were computed using coancestry, inbreeding (F), and contribution (CFC) programs. 16
Ethics committee approval for treatment of animals was not required, as all samples used in this study had been collected by veterinarians for routine purposes, and were reused for the research presented here.
Selection and genotyping of markers
A total of 54,001 SNPs were screened using the Illumina Bovine SNP50K BeadChip.
17
SNPs were analyzed using an Illumina Bead-Station 5.2 genotyping instrument (Illumina Inc.), and genotyping was performed using BeadStudio 3.0 (Illumina Inc.) software. SNPs were removed on the basis of the following criteria: 1) Markers were filtered to exclude loci assigned to unmapped contigs, or unpositioned according to the latest reference assembly of the bovine genome Btau 4.0, and markers on the X and Y chromosomes. 2) Monomorphic SNPs and those with minor allele frequency smaller than 0.05 were filtered, because it is known that LD between SNPs with a low minor allele frequency is biased upwards, and thus high-frequency polymorphisms are preferable for accurate estimation of LD.
18
3) Animals with genotype completeness smaller than 90% were excluded. 4) Markers with significant departure from H-W equilibrium (
Haplotype phase reconstruction
Haplotype phases were inferred from pedigree by a localized haplotype clustering model (LHCM) method, 19 which is a family rule (Mendelian segregation and linkage)-based algorithm. Only maternally inherited haplotypes were used for further detailed analyses, in order to minimize the effect of over-representation of paternally inherited haplotypes within pedigrees of the bulls.
SNP annotation information
Annotation information was based on genomic positions of
Measures of linkage disequilibrium
We used the GOLD 1.1.0 program
21
to construct LD maps from the maternally inherited haplotypes, thus avoiding the over-representation of paternally inherited haplotypes within the primarily male pedigrees. GOLD computes Lewontin's disequilibrium coefficient
Past effective population size using LD
To observe the past effective population size (
Long-range haplotype (LRH) test
Under neutral evolution, positive selection is expected to accelerate the frequency of an advantageous allele faster than recombination can break down LD at the selected haplotype. 27
The LRH test calculates the relative extended haplotype homozygosity (REHH), and assesses the significance of REHH by use of simulations, as described in Sabeti et al.
8
In brief, core regions (a pair of SNPs to be in strong LD if the upper 95% confidence bound of
Results
The filtering resulted in 35,968 useful SNPs, which were used for further analysis. More precisely, 3,366 SNPs (6%) did not produce any genotypes, Of the initial SNPs, 7,814 (14.5%) were monomorphic, and 5,953 (11%) were filtered out because of low minor allele frequency (MAF) (<0.05). The fraction of SNPs excluded because of partially (>25%) missing genotypes and deviation of Hardy-Weinberg equilibrium (HWE) was negligible (185 SNPs, <0.1%). This subset of markers covers 2,543.6 Mb of the genome with 70.57 ± 69.0 kb average adjacent marker spacing. The largest gap between SNPs (2081.5 kb) was located on chromosome 10. For the SNPs analyzed in this study, the average observed heterozy-gosity was estimated at 0.37 ± 0.12. The average MAF of all SNPs before quality control (QC) was 0.20, and using the filtered SNPs, the averaged MAF increased to 0.27, which was higher than the average MAF of SNPs reported on the Illumina Bovine SNP50K BeadChip for Hanwoon (0.198). 29 The SNPs genotyped showed an almost uniform distribution across the common frequency classes. This is probably due to the design of the SNP array, which was optimized with respect to a uniform SNP spacing and MAF distribution (Fig. 1). 18

Minor allele frequency of SNPs.
In total, 1,760 core regions were identified, incorporating 5,915 SNPs which corresponded to 7.72% of the combined length of all the autosomes (Table 1). The mean core region length was estimated at 111.53 ± 71.89 kb, with a maximum of 2,081.46 kb. There were 141 core regions spanning 161.02 Mb on chromosome 1, and 20 core regions covering 51.90 Mb on chromosome 29. These were the largest and smallest haplotypic structures in the genome. An additional 1,318 core region consisting of three SNPs were identified (Fig. 2). The maximum number of SNPs in a core region was 14 from chromosome 14. There were 326 core regions with four SNPs, 82 core regions with five SNPs, 18 core regions with six SNPs and 8 core regions with seven SNPs.

(
Genome-wide summary of marker and core region(CR) in Hanwoo cattle.

Linkage map, NCBI Map Viewer (http://www.ncbi.nih.gov/mapview).
The proportion of total core region lengths on chromosome length.
Number of SNPs forming core regions.
Linkage disequilibrium
Decay of LD (
Pairwise linkage disequilibrium (

Percentage of pairs of SNPs with
Percentage of pairs of SNPs with
Variation of LD in different distance bins of individual chromosomes combined over the genome was examined (Fig. 3). For marker pairs 0–40 kb apart, the average
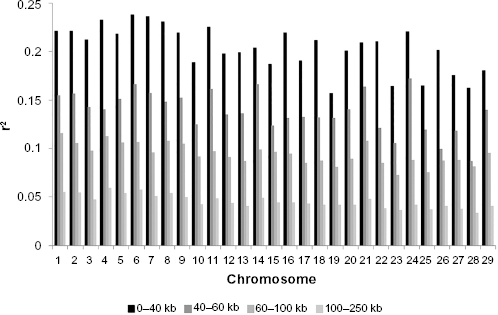
Chromosome-wise average
Linkage disequilibrium (
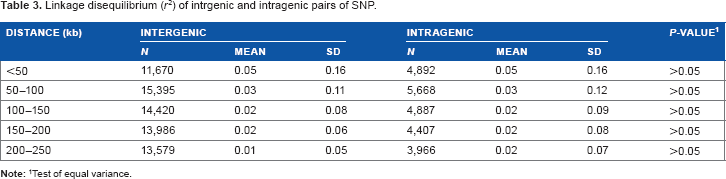
Test of equal variance.
Effective population size
Past effective population size was estimated chromosome-wise using pairwise SNP

Estimated effective population size over the past generations from linkage disequilibrium data. The variability at each point time reflects the variation of estimates between the 29 autosomes.
Effective population size(Ne) in the past Hanwoon population.
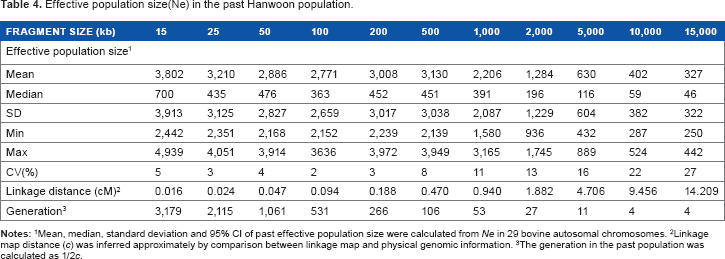
Mean, median, standard deviation and 95% CI of past effective population size were calculated from
Linkage map distance (
The generation in the past population was calculated as 1/2
Whole-genome screening for selection signatures
For all 1,760 core regions, a total of 12,186 LRH tests with an average of 6.92 tests per core region were made. Taking this into consideration, we skipped core haplotypes with frequency <25% and plotted the -log10 of the

Genome-wide map of –log10(REHH
Summary statistics of whole genome extent haplotype homozygosity test.
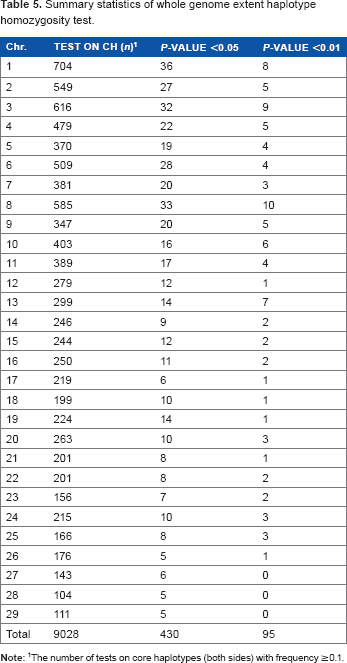
The number of tests on core haplotypes (both sides) with frequency ≥0.1.
We also explored a quantitative trait loci (QTL) database available online (http://www.animalgenome.org/QTLdb/cattle.html) 30 to identify any overlapping of the outlying core regions with published QTL in dairy and beef cattle. Additional File 1 lists the traits, approximate position, and reported population of the overlapping QTL for each core region.
Discussion
The pattern of LD in domestic animals means that a marker may be in LD with a QTL some distance away and show an association with the trait affected by the QTL; it also tells us how natural selection, genetic drift, recombination, and mutation all affect the levels of LD.2,31 In this study, we present an analysis of LD of 35,968 SNP genotypes in Hanwoo, in which only maternally inherited haplotypes were used. We elected to use maternal haplotypes in consideration of the complexity of the pedigrees, with dams contributing information to multiple families. Bohmanova et al reported that the use of maternal haplotypes is recommended for analyses of LD in populations consisting of large paternal half-sib families. 32
Comparable estimates of extensive LD have been reported in cattle.18,32,33 Consistent with previous analysis in cattle, the decline of LD as a function of distance was rapid, with average
There were no differences in the extent of LD and the decline of LD with the distance between intragenic and intergenic regions (Table 3). Indeed, knowledge of LD with genes is important. However, noncoding elements such as miRNAs might also play a role in many inherited traits. Therefore, these results suggest that noncoding regions have been an important substrate for adaptive evolution in Hanwoo.
The number of SNPs needed depends on the distance over which LD operates. If SNPs are too far apart, a QTL may not be in sufficient LD with any of the markers, and so will be undetected.
2
Meuwissen et al reported the required level of LD (
The pattern of LD observed in a population depends on the history of the population, especially the history of its effective population size. 2 This study shows a persistent decline in the effective population size. In particular, there are sharp declines at two time intervals and one distinctive time point (Fig. 4; Table 4). The distinctive point at 100 generations ago shows a sharp decline in population size, which is consistent with previous studies.39,40 This pattern can be explained by breed formation and modern breeding programs. 15 The first sharp decline of the effective population size was observed in our study ~25–50 generations ago, and is due to enhancement of selection. A second and more recent sharp decline seems to have occurred ~10–25 generations ago and might thus correspond to several events related to the intensive use of artificial insemination (AI). Overall, the Korean national economy has grown very rapidly since 1970, which has brought about a change in the Korean lifestyle, including significant consumption of meat from cattle. In this regard, improvement of selection methods, together with the adaptation to different agro-ecological constraints, has been necessary and might have had a direct effect on the population structure of cattle.
The estimated effective population size for the most recent time is around 300 individuals. Compared to other reports,18,33,39–41 recent
Positive selection is expected to accelerate the frequency of an advantageous allele faster than recombination can break down LD at the selected haplotype. 27 In this report, we employed the LRH test by selecting a “core” haplotype with elevated EHH relative to other core haplotypes, at the locus conditional on haplotype frequency. This was used to identify selection signatures within a single population. Our result revealed 95 regions exhibiting the footprint of recent positive selection at a threshold level of 0.01.
We found an overlap of the 11 core regions presenting top
Eleven QTL regions, associated not only with production traits but also with meat and carcass traits, have been identified. Within the last century, Hanwoo have been intensively selected for both the quality (marbling, tenderness, and flavor) and the quantity (carcass weight) of meat to meet the growing demand for beef in Korea. Cattle farming methods have changed dramatically, so it is not surprising that QTLs identified are related to meat traits, showing the signatures of recent selection. Especially for BTA14, which harbors known genes and QTL for several economically important traits, we found agreement between the regions (24.3–25.4 Mb) and those that had previously been identified to harbor carcass weight QTL peak on Hanwoo.
46
We extended core regions in both directions up to 1 Mb as the length of the core domains. We identified
Conclusion
In this work, the Illumina Bovine SNP50K chips were used for SNP genotyping in an elite Hanwoo population comprising 61 sires and their 486 steers. Haplotype phases were inferred by an LHCM method, and maternal haplotypes were chosen across all autosomal chromosomes.
To identify selection signatures on any genomic position, an LRH test was applied. Our results revealed 95 regions exhibiting footprints of recent positive selection (
Author Contributions
Conceived and designed the experiments: YL, JJK. Analyzed the data: YL, JJK. Wrote the first draft of the manuscript: YL. Contributed to the writing of the manuscript: YL, JJK. Agree with manuscript results and conclusions: YL, JJK. Jointly developed the structure and arguments for the paper: YL, JJK. Made critical revisions and approved final version: YL, JJK. Both authors reviewed and approved of the final manuscript
Supplementary Material
Supplementary Table 1.
Summary statistics for 11 core haplotypes showing the lowest
Footnotes
Acknowledgments
Thanks for the academic editor and reviewers for their comments, which were very helpful in improving the manuscript.