
Abstract
Knowledge of markers in the human genome which show spatial patterns and display extreme correlation with different environmental determinants play an important role in understanding the factors which affect the biological evolution of our species. We used the genotype data of more than half a million single nucleotide polymorphisms (SNPs) from the data set Human Genome Diversity Panel (HGDP-CEPH-CEPH) and we calculated Spearman's correlation between absolute latitude and one of the two allele frequencies of each SNP. We selected SNPs with a correlation coefficient within the upper 1% tail of the distribution. We then used a criterion of proximity between significant variants to focus on DNA regions showing a continuous signal over a portion of the genome. Based on external information and genome annotations, we demonstrated that most regions with the strongest signals also have biological relevance. We believe this proximity requirement adds an edge to our novel method compared to the existing literature, highlighting several genes (for example DTNB, D0T1L, TPCN2, RELN, MSRA, NRG3) related to body size or shape, human height, hair color, and schizophrenia. Our approach can be applied generally to any measure of association between polymorphic frequencies and continuously varying environmental variables.
Introduction
From an evolutionary point of view, human biological variation can result from natural selection, genetic drift and demographic processes. In human population genetics, several ways have been found to highlight genes that may be subject to selective pressures, and in recent years whole genome scanning techniques have made it possible to find signatures of selection.1–4 The Human Genome Diversity Project (HGDP-CEPH) database 5 has been repeatedly investigated in order to identify markers in the human genome which show geographical patterns and to explain how different selective forces can shape human genetic variations across continents. One strategy for the detection of spatial selection signatures is the outlier approach.2,6,7 Using genome-wide data sets genotyped in different human populations, genetic variables–-such as single nucleotide polymorphisms (SNPs)–-that exhibit extreme correlations with latitude or with other environmental determinants are identified as candidate targets for selective pressure. By “extreme correlation” we mean that the value of a certain statistic, measuring the strength of the relationship between allele frequencies and latitude or other environmental variables, falls in the tails of the distribution of the same statistic over the whole genome. Many choices are possible for the relevant statistic, ranging from a simple (either Pearson or Spearman) correlation coefficient between the latitude and the frequencies of either one or two alleles of a SNP to a Bayes factor comparing two models that do and do not, respectively, take into account the effect of a dichotomous environmental variable on the distribution of a genetic variant. From a technical point of view, the outlier approach is just a reformulation of the concept of statistical significance, ie, variation with respect to a reference distribution.
The outlier approach has been used to study sodium homeostasis balance as an example of adaptation. In hot and dry climates, genes influencing salt and water retention are favored by selection, explaining in this way large inter-ethnic differences in the prevalence of salt-sensitive hypertension.8,9 Other important research has been conducted to assess the correlation between four variables that summarize climate and the frequencies of 873 tag SNPs in 82 genes related to energy metabolic pathways. 6 The outlier approach has also been used to demonstrate that allele frequencies of a subset of genes coding for blood group antigens vary with levels of pathogen richness, supporting the idea that these loci affect susceptibility to infectious diseases. 10 This finding, which is compatible with previous evidences on the correlation between HLA class I diversity and pathogen richness, 11 is important for stressing the role of diseases and pathogens, like virus protozoa fungi, in shaping human variations. 12 Finally, a very comprehensive article on the HGDP-CEPH database (enriched with the Hap Map and other human populations databases) has recently been published, in which the outlier approach is used to highlight polymorphisms and pathways correlated with ecoregion membership and diet. 13
Our idea is to reinforce the outlier approach by considering a criterion of proximity between significant variants. In the search for targets of selective pressure, we believe it is important to focus on those DNA regions which repeatedly contain values which are labeled as significant by the outlier approach. In other words, we look for evidence of a continuous signal over a portion of the genome which can strengthen the significance of a cluster of markers labeled as significant by the outlier approach alone and we built statistical tools.
In this paper we therefore adopt a search-and-confirm approach which integrates the outlier approach by identifying regions of the genome where not just one, but a significant number of SNPs are located in the tails of the distribution of the relevant statistic, when compared to the number of SNPs originally genotyped in the same region. This is done in the following three steps, which are further illustrated in the complete workflow process diagram in Figure 1:
The outlier method: We identify 1% significant SNPs as having an absolute value of the Spearman correlation coefficient with latitude above its 99th percentile; The proximity-based algorithm: Using the methods described in detail in the Materials and Methods section, we select candidate regions in the genome which exhibit the strongest signals, ie, the regions where the significant SNPs identified above are present at a significantly higher rate when compared to the number of originally genotyped SNPs; Biological relevance: We investigate the biological relevance of the strongest signals by comparing our data with results from Genome Wide Association studies (GWAs),
14
by studying the canonical pathway processes through gene-annotation enrichment analysis
15
and by comparing our analysis with previously published genomic scans for selective sweep.3,16

Graphical workflow process for the study.
Materials and Methods
We describe here our methods with reference to the three-step process described in the Introduction.
Step 1 : Our data and the outlier method
We used a data set of 660,832 SNPs genotyped in 51 human populations distributed worldwide from the HGDP-CEPH panel. 1 As underlined by a previous article, 17 within the HGDP-CEPH panel there are some closely-related individuals; in order to overcome this possible source of bias we excluded one member of each relative pair and we used 938 HGDP-CEPH individuals. Information about sample sizes and latitudes of the populations can be found on the CEPH homepage http://www.cephb.fr/en/hgdp/table.php. 5 Only 22 autosomes are included in our analysis; we also removed SNPs with more than 10% of missing genotypes and the ones that failed the Hardy-Weinberg equilibrium test in at least one population. After filtering, we use 545,209 SNPs.
Statistical analysis is performed using R. 18 We calculated Spearman's correlation (the correlation coefficient between the ranks of two variables) between absolute latitude and one of the two alleles of each SNP and, using the outlier approach, we identified those SNPs which have an absolute Spearman's correlation coefficient falling in the upper 1% tail of the distribution (Fig. 2).
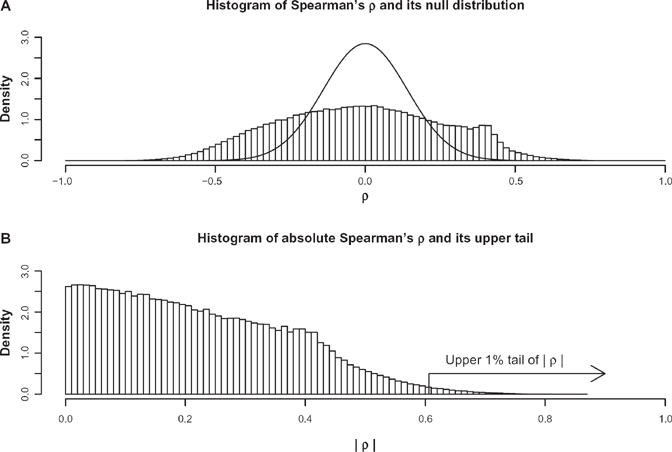
(Panel A) Histogram of the values of Spearman's correlation coefficient over all the SNPs and theoretical approximate density of the Spearman's correlation coefficient under the hypothesis of population null correlation. (Panel B) Histogram of the absolute values of Spearman's correlation coefficient over all the SNPs. Using the outlier approach, we identify significant SNPs in the 1% upper tail of this distribution.
Step 2: The proximity-based algorithm
For each chromosome, we now have two sequences of serial positions: one for all genotyped SNPs and one for the significant SNPs, the latter of which are included in the former. Each chromosome is indexed by the sequence of base pairs: as an approximation, we can view a chromosome as a linear segment and the position of a SNP as a point of that linear segment. Based on the two sequences of points, we can define two cumulative counts depending on a generic point l, known in statistics as counting processes:
S(l) = number of SNPs with a position smaller than or equal to l S.01(l) = number of significant SNPs with a position smaller than or equal to l
with l varying from 1 (the first bp in the chromosome) to the position of the last bp of the chromosome. As an example, the two counting processes are plotted for chromosome 1 in Figure 3. Cumulative counts are a convenient way to compare the incidences of the different kinds of SNPs over different genomic regions (a simple dot plot would not do it, due to the sheer number of SNPs involved). If, over a certain segment of the chromosome, there is a greater-than-usual incidence of significant SNPs, then the relative increment of S.01(l) over that segment will be greater than the relative increment of S(l) over the same segment. In other words, the graph of the S.01(l) counting process will be steeper than S(l), up to a proportionality factor. Our proposal is to identify those genome regions which exhibit extreme concentrations of outlying SNPs.

Counting process representation of the location of the candidate regions of chromosome 1.
We could formalize this search as a change-point problem for counting processes: in certain intervals to be estimated, the intensity of the S(l) point process–-a function modelling the instantaneous rate of incidence of the process–-would be higher than in other regions. Due to the size of the problem and to the approximate nature of our search- and -confirm approach, we prefer a simpler proximity-based algorithm as follows.
For each pair of significant SNPs located at points l1 and l2, with l1 < l2 on the chromosome, we define
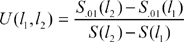
As a technical note, it would probably be a good idea to penalize large windows, for example by dividing the U(l1, l2) statistic above by a penalty term (l2–l1)g with g equal to some number between 0 and 1. The final results would not change a lot (results not shown) and it would be difficult to commit to a specific g; therefore we decide to use the U(l1, l2) statistic without a penalty term.
For each chromosome and for each significant SNP in position l1, we computer U(l1, l2) for each of the other significant SNPs in position l2 within a distance of 1000 Kb from the original one. This is done to reduce the problem to a manageable size, under the assumption that relevant proximities are smaller than 1000 Kb.
We built the new reference distribution of all U(l1, l2) values over all chromosomes, excluding from the analysis all U(l1, l2) values relative to intervals (l1, l2) which included fewer SNPs than a threshold s, which has been chosen to be equal to 3 in this work. This is done to avoid very high automatic values of U(l1, l2) when two significant SNPs happen to be adjacent. We selected the first 1000 SNPs contained in regions corresponding to the highest U(l1, l2) values. A fixed number, rather than a fix tail area, was chosen to facilitate the discussion of the robustness of our method to varying parameters (see end of section Results).
Step 3: Biological relevance of the strongest signals
To accomplish step 3 as outlined in the Introduction, we proceeded to the biological cross-validation of our findings, which insofar had been based mainly on statistical grounds. We focused on the genes tagged by the SNPs we found, since our goal was to detect continuous signals coming from proximal groups of SNPs belonging to the same gene. To link our findings to the results of genome wide data, we first compared our gene list with the June 2012 update of the Catalog of Publish ed GWAs. 14 Next, we scanned our gene list using a bioinformatic enrichment tool named Genecodis 2.0 15 to obtain a summary of the most enriched biological processes or pathways. Finally, we compared our analysis with previously published genomic scans for selective sweep in order to find possible overlaps in signals.
Results
We calculated Spearman's correlation between absolute latitude and one of the two alleles of the SNPs found in the HGDP-CEPH panel and, following the outlier approach, we identified those SNPs which have an absolute Spearman's correlation coefficient falling in the upper 1% tail of the distribution. The histogram of Spearman's correlations ρ's is plotted in Figure 2A. Its null distribution for 51 pairs of numbers has been overlaid on the same graph (Fig. 2A). It is a normal distribution with variance 1/50 due to a well-known result. 19 The discrepancy between the two distributions is due to SNPs which are correlated with latitude for reasons other than chance alone, for example due to environmental selection factors. Following the outlier approach, the upper 1% of the distribution of the absolute value of ρ, corresponding to |ρ| > 0.606, is identified in the histogram of the absolute value of ρ (Fig. 2B). It corresponds to 5452 outlying SNPs in the tails of the ρ distribution.
The candidate regions and the annotations emerging from the application of Step 2 described in the Introduction are contained in Additional 1 in the online supporting information. As an example, candidate regions which were identified in chromosome 1 are shown in Figure 3. The 1000 top SNPs emerging from the proximity-based algorithm enabled us to identify 467 intergenic and 533 genie SNPs, harboring 146 genes. We found 23 coding non synonymous (NS) changes and 6 coding synonymous changes. 372 were intronic and 107 were on the mRNA 3′UTR.
Finally, we gathered the biological knowledge of the strongest signals by comparing them to the Catalog of Published Genome-Wide Association Studies updated to June 2012. The genes which appear on this Catalog and additionally appear in candidate regions according to our proximity-based algorithm, are shown in Additional file 2. A short list of the most interesting signals are shown in Table 1. Several genes shown in that table are associated with metabolism-related phenotypes (like celiac disease for IL21 interleukin 21, Gene id 59067) 20 and adiposity (MSRA Gene id4482) or variants associated with hair color in Europeans, like TPCN2 gene (two pore segment channel 2, gene ID 219931) 21 and several with schizophrenia. At the same time, we compared our gene list with genes reported in OMIM. Several of our genes which show a correlation with latitude also implied some traits. For example, DOT1L gene (DOT1-like, histone H3 methyltransferase Saccharomyces cerevisiae) gene ID 84444 is associated with height 22 or DTNB gene dystrobrevin, beta ID 1838 which is affecting adult human height. 23 A complete table with the genes reported also in OMTM Disease database is in Additional file 3.
List of several genes reported in previously published GWAs and showing continuous correlation signals with our proximity based method.

We analyzed Kyoto Encyclopedia of Genes and Genomes pathways (KEGG) using as reference set all genes in the Entrez-gene database and, as a statistical test, the hypergeometric one with a Benjamini-Hochberg correction for multiple testing at significance level equal to 0.05. Several KEGG pathways reached significance. The first was the extracellular matrix (ECM) receptor interaction (KEGG number: hsa04512) for the following genes: RELN reelin gene ID 5649; ITGB6 integrin beta 6 gene ID 3694; COL6A3 collagen, type VI, alpha 3 gene ID 1293. This pathway reaches a raw P-value of the hypergeometric test equal to 0.0011 and a P-value adjusted for multiplicity around 0.01. In order to look for overlaps with scans of the human genome for signals of positive natural selection, we compared our results with SNPs with significant composite of multiple signals (CMS) but only one intersection was found between the two gene lists concerning rs2256670 and rs2711853 both on RELN reelin, gene ID 5649. 16 A variety of choices were made in the actual implementation of the proximity-based algorithm described in Step 2 in the previous section. The two most important parameters set to reasonable values are (a) the maximum distance over which we search, which is set to 1000 Kb in Step 2, and (b) the minimum number of consecutive SNPs required, which is set to 3 in Step 2. In order to study the robustness of our method with respect to different values of these parameters, we varied the maximum distance and noticed (not shown) that the results where unchanged for distances down to 100 Kb.
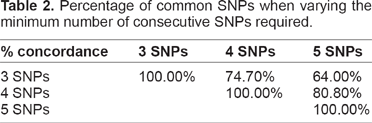
The algorithm is instead sensitive to the minimum number of consecutive SNPs required: if we increase it from 3 to 5, for example (it would not make sense to consider a minimum much higher than 5), different SNPs and regions turn out to be significant, as shown in Table 2. For example, the number of selected SNPs shared when applying a minimum of 5 and when applying a minimum of 3 is 64%. This made us consider what would happen for varying this threshold. The changes are not dramatic (Table 2) but some interesting genes, like AGT, ADCY9 and WWOX would come out from the analysis with a threshold equal to 5.
Percentage of common SNPs when varying the minimum number of consecutive SNPs required.
Discussion
In this paper we examined the HGDP-CEPH data again by integrating the outlier approach with a novel proximity-based algorithm.
Only latitude was used for ecological conditions, rather than using a multiplicity of variables as in Hancock et al 6 for example. We made this choice for the sake of simplicity, since latitude is correlated with different variables like short wave radiation flux, mean winter and summer temperatures, rainfall and pathogen richness. It should therefore provide a good proxy for the selective pressures that shaped variation in our genome. Even though we use a simple correlation measure such as Spearman's ρ with latitude only, we emphasize that the resulting signal should be a continuous and persistent proportion of background information, represented by all originally genotyped SNPs. We believe this proximity requirement adds an edge to our novel method when compared to existing literature. Our approach is applicable to any measure of association between polymorphic frequencies and environmental variables. It could be applied, for example, to complex statistics such as the minimum rank statistic, based on Bayes Factors and on rank transformations, of Hancock et al. 13
With our method we identified different genes, some of them already reported in the literature, dealing with different traits or diseases. GWAs include the scanning of all or most of the genes of different individuals aimed at finding susceptibility loci for traits or diseases. GWAs, so far, have allowed the identification of more than 7688 associated SNPs in humans. We compared our list of genes with GWAs results. Some interesting signals can be pointed out, for instance the correlation between skin pigmentation and latitude. It is well known that two coding variants in TPCN2 are associated with hair color in Europeans. 21 At the same time MSRA (methionine sulfoxide reductase A gene) is related to the melanin formation in the hair follicle melanocyte. 24 Remarkably, MSRA gene is also related to schizophrenia25,26 but also with adiposity 27 and hypertension. 28
Several other genes in our list (see Additional file 1) can be associated with vitamin D related genes, known to show a latitude driven cline. 7 An example is SMARCA2, (SWI/SNF related, matrix associated, act in dependent regulator of chromatin, subfamily a, member 2), described as a component of a human multiprotein complex that that interacts directly with the vitamin D receptor. Schizophrenia genes are correlated with latitude and in our list several schizophrenia genes appear, like GRID1,29,30 MAGI2, 31 NRG3, 32 NRXN3, 33 RARB and RELN. 34
Region CYP19A1 in our list is known from GWAs to exhibit association with adult height35,36 whose distribution is related to latitude. Two more genes in our list, DOT1-like, histone H3 methyltransferase (S. cerevisiae)22,35 and dystrobrevin, beta 23 are reported in OMIM to be related with height.
Several others genes are related to Celiac Disease (CD) which strongly correlates with latitude. Infectious agents are implicated in the pathogenesis of many autoimmune diseases like CD. This observation may imply that there is a relationship between one or more infectious agents, latitude related environmental exposure to gluten and others genetic susceptibility loci, and the development of this disease. For a complete review see Plot and Amital, 2009. 37 The RUNX3 gene and IL21, in our list, are implicated with CD. 38 In the same paper, another gene FRMD4B previously known as GRSP1, appearing in our Table 1 is also associated with CD. 38 RUNX3 gene is also required for CD8 T cell development during thymopoiesis. 39
One of the most interesting genes highlighted by our work is ANK2 (ankyrin 2, neuronal) which is implicated in cardiac arrhythmias due to abnormal variations in QT interval. 40
Finally, the enrichment of genes in the KEGG pathway called extracellular matrix (ECM) receptor interaction (KEGG number: hsa04512) is note worth because these molecules are exploited by a number of pathogenic micro-organisms as receptors for cell entry. This can be interpreted as a signal of different forces played by pathogens on living cells in different environments.
Conclusions
Our study complements the growing body of knowledge surrounding scans for natural selection in humans using a method that uses the proximity criterion in addition to the outlier approach. Our findings support the hypothesis that latitudinal genetic diversity gradients are present in humans and reflect genetic adaptations to different environmental pressures that have shaped the human genome.
Authors' Contributions
CDG designed the biological rationale for the study and MG provided the statistical tools to implement it, both authors wrote and revised the manuscript. MU and AP performed the data analysis. GM revised the manuscript. All authors read and approved the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgements
The authors thank the Human Genetic Foundation (HuGeF) Laboratory of Genomic Variation in Human Populations and Complex Disease group for biological and bioinformatics discussion.
Additional Files
Additional file 1 in the online supporting information contains all regions selected by the proximity-based method, duly annotated.
Additional file 4 in the online supporting information contains R scripts to perform the necessary calculations.