
Abstract
The adaptive evolution and coevolution of the ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (rbcL) gene in the genus Hildenbrandia were studied based on phylogenetic tree construction and the physicochemical properties and the secondary structures of protein encoded by rbcL (Rubisco large subunit) were analyzed. The amino acids compositions and grand average of hydropathicity of freshwater H. rivularis and marine H. rubra were similar. Rubisco large subunit of Hildenbrandia was hydrophilic and the secondary structure was primarily composed of α-helixes and β-sheets, revealing the relatively stable structure of this protein. The predicted phosphorylation sites in H. rivularis and H. rubra were 33 and 36, respectively. No positive selection sites were detected in the genus Hildenbrandia, implying that rbcL gene evolved either neutrally or under purifying selection. A total of 41 coevolutionary groups were detected in the Rubisco large subunit of Hildenbrandia and the coevolving sites are in closer proximity in 3-dimensional structure of the protein. Despite the long evolutionary history, rbcL gene in genus Hildenbrandia under different environments is rather conservative.
Keywords
Introduction
Rhodophyta are a monophyletic lineage in Archaeplastida and also well known for their contribution to algal evolution as a plastid donor to the chlorophyll-c containing eukaryotic groups through secondary endosymbiosis.1,2 Red algae primarily inhabit in marine environment and only a small portion (less than 3% of the over 6500 species) occur in freshwater habitats.3,4 They consist of 7 classes and the class Florideophyceae includes both marine and freshwater taxa, thus making it a suitable model for studying environmental adaptation. Genus Hildenbrandia belong to Rhodophyta, Florideophyceae, Hildenbrandiales, which is distributed globally in both marine and freshwater habitats, and is characterized by simple crustose thallus construction with a single basal layer and derived vertical cell files. The marine and freshwater species are similar in morphology and it has been proposed that freshwater Hildenbrandia might have arisen by invasions of marine Hildenbrandia into freshwater habitats. 5 Among this genus, H. rivularis inhabited exclusively in freshwater and H. rubra is distributed in marine environment. 6 The freshwater Hildenbrandia populations are only distributed in streams with high water temperatures (mean value = 21°C) and high specific conductance values (mean value = 223 μS cm−1), thus they are strict in habitat selection. 7 Freshwater Hildenbrandia is increasingly rare in relation to the serious pollution of water environment. Investigation on its adaption to freshwater environment is needed urgently.
The rbcL (1, 5-ribulose bisphosphate carboxylase/oxygenase large subunit) gene encodes large subunit of rubisco protein, which is a critical enzyme for both the reductive and oxidative photosynthetic carbon cycles. It has a decisive influence on the net photosynthetic rate and is the primary enzyme responsible for autotrophy of plant cells.8 -10 The level of amino acid substitution in rbcL was related with the absence or presence of pyrenoids in green algae. 11 Therefore, the evolutionary changes related to Rubisco’s function were likely subjected to selection pressures owing to its direct relationship to the biological fitness of the plant. Genus Hildenbrandia inhabit in both marine and freshwater enviroments.5,6 The environmental stresses such as oxidative stress and osmotic stress have been proved to affect the activity of RuBisCO. 12 Additionally, trends of higher evolutionary rates within protein-coding genes in freshwater taxa compared with marine relatives have been observed in previous literature. 13 The adaptive evolution of the chloroplast gene revealed by molecular evidences has been reported on several plant lineages. Adaptive evolution analysis on rbcL genes of xerophytic Pteridaceae ferns was conducted and 3 positive selection loci playing prominent roles in maintaining Rubisco function were detected. 14 By contrast, the adaptive evolution of the rps4 gene in ferns was studied and no positive selection site was detected revealing the stabilized structure and function of this gene. 15 Research on the adaptive evolution of red algae were scarce, except for the adaptive evolution analysis on rbcL gene in Batrachospermales. 16
The accumulation of rbcL sequences in public databases and advances in computing technologies offer the opportunity to analyze the evolution of this gene. The PAML software package was developed to detect the presence of positive selection sites by calculating the ratio (ω-value) of non-synonymous (dN) and synonymous substitution (dS) of amino acid sequences. The ratio indicates negative purifying selection (0 < ω < 1), neutral evolution (ω = 1), and positive selection (ω > 1). 17 It is common that the coevolution of amino acid sites occurred within proteins. Adaptive evolution analysis complemented with the identification of coevolutionary dynamics can highlight the intricate co-adaptive relationships between residues in the protein. Dynamic coevolution between functionally relevant sites were considered necessary for understanding the evolutionary process of proteins. 18
In this study, adaptive evolution and coevolution analysis on the rbcL gene of Hildenbrandia were analyzed. Three-dimensional structure of the Rubisco protein and the coevolution of amino acids in the protein were illustrated. We aim to explore the genetic changes and the adaptability of protein functions of red algae under different environmental pressures (marine vs freshwater environments).
Materials and Methods
Phylogenetic tree construction
A total of 26 rbcL gene sequences and 3 outgroup DNA sequences were downloaded from GenBank (Supplemental Table S1). The sequence dataset was aligned using ClustalX software. 19 The unaligned bases at both ends were cut to obtain 29 sequences of the same length, each of which was composed of 321 codons.
The DNA sequences were translated into amino acids using MEGA7.0 software, 20 and the optimal evolutionary model of the sequence dataset was computed using ProtTest software 21 (Supplemental Table S2). Maximum likelihood method (ML) was used to build the phylogenetic tree (repeating the calculation for 1000 times) using PhyML3.0 software.22,23
Physicochemical properties and the secondary structures of protein prediction
The amino acid sequences of H. rivularis (accession number KX284723) and H. rubra (accession number KX284724) were used as references to predict the protein hydrophilic/hydrophobic values in ProtScale (http://web.expasy.org/protscale/). Amino acid scale was set as Hphob./Kyte & Doolittle and weight variation model was linear. The physical and chemical properties of the selected rbcL protein-coding genes were identified using the Prot Param (http://web.expasy.org/protparam/). The same sequences were submitted to NetPhos 3.1 Server (http://www.cbs.dtu.dk/services/NetPhos/) to predict the protein phosphorylation sites. All 3 residues including serine, threonine, and tyrosine were predicted. Sites with score value higher than 0.5 were considered as positive prediction. The secondary structures of the proteins were analyzed using the Protean module in the package DNASTAR.Lasergene.v7.1 software. Two methods were used including Garnier-Robson and Chou-Fasman. The former method was based on the calculation of the possibility of a specific amino acid residue within a specific structure and Chou-Fasman method was based on the crystal structure of amino acid residues.24,25
Adaptive evolutionary analysis
Based on the phylogenetic tree constructed from 29 Hildenbrandia sequences, the positive selection analysis was conducted using codeml model in PAML4.9 software package. 17 The branch model, site model and the branch-site model were used for detecting the positive selection sites within rbcL sequences among lineages. The branch model in PAML assumes the ω value varies in different branches, with 1 ratio (model = 0) assuming that all branches have been evolving at the same rate, the 2 ratio (model = 2) allowing the foreground branch to evolve under a different rate with the background branch, and free ratio (model = 1) assuming that all branches evolving under various rates. 26 The statistical likelihood ratio test (LRT) was used to test the reliability of each model with the 1 ratio as null model and the 2 ratio and free ratio as alternative models. 27 The site models allow ω to vary in each site (codon) within the gene and were mainly used to detect whether positive (ω > 1) and negative selection (ω < 1) sites exist in the rbcL gene. 28 LRT was used to compare 3 pairs of models including M1a (near-neutral evolution) and M2a (positive selection), M0 (single ratio) and M3 (discrete ratio), M7 (beta) and M8 (β & ω), with the former as null hypothesis and the latter as the alternative hypothesis. 29 The M7 model is a null model that allows 10 classes of sites with a ω beta-distribution within the interval 0 ⩽ ω ⩽ 1 and the alternative M8 model adds an eleventh class of sites with ω > 1. The LRT compares twice the log-likelihood difference between the 2 models of each pair to the χ2 distribution. If the twice log-likelihood difference is above a critical χ2 value, then the null model is rejected, and the positive selection is statistically significant. In the branch-site model, LRT was conducted to compare ModelA null (ω was set as 1) and ModelA (allow sites to be under positive selection on the foreground branch).
Amino acid sequences of accession number KX284723 and KX284724 were uploaded to the Swiss Biotechnology Research Institute (European Bioinformatics Institute: https://swissmodel.expasy.org/) to predict the Rubisco large subunit 3-dimensional structure based on homology modeling. 30
Identification of intra-protein coevolutionary pattern
The evolutionary dependencies were analyzed to identify functional/structural dependencies among residues in the RbcL subunit based on the analyzed 3-dimensional structure of Rubisco large subunit (PDB ID: 1BMV). Intra-protein co-evolution of amino acid sites encoding by rbcL gene of Hildenbrandia was analyzed by the parametric test method, 31 mutual information method, 32 and Pearson correlation coefficient method of CAPS (Coevolution Analysis Using Protein Sequences) software.33,34
Results
Physicochemical properties and the secondary structures of protein prediction
The amino acid compositions of each Rubisco large subunit in H. rivularis and H. rubra were similar, only different in numbers of alanine (Ala), arginine (Arg), Asparagine (Asn), Isoleucine (Ile), Lysine (Lys), Methionine (Met), Proline (Pro), Serine (Ser), Threonine (Thr), and Tyrosine (Tyr) (Figure 1a). The grand average of hydropathicity (GRAVY) was calculated to measure the protein hydrophilicity/hydrophobicity. Hydrophobicity is revealed by a positive GRAVY value while hydrophilicity is revealed by negative value. The GRAVY values of the Rubisco large subunit measured by Prot Param were −0.134 for H. rivularis and −0.132 for H. rubra, thus indicating the protein is hydrophilic and water-soluble. The GRAVY values of the amino acid sequence of Rubisco large subunit of both H. rivularis and H. rubra were the smallest (−2.500) at the 305th site, indicating that the site is extremely hydrophilic (Figure 1b).
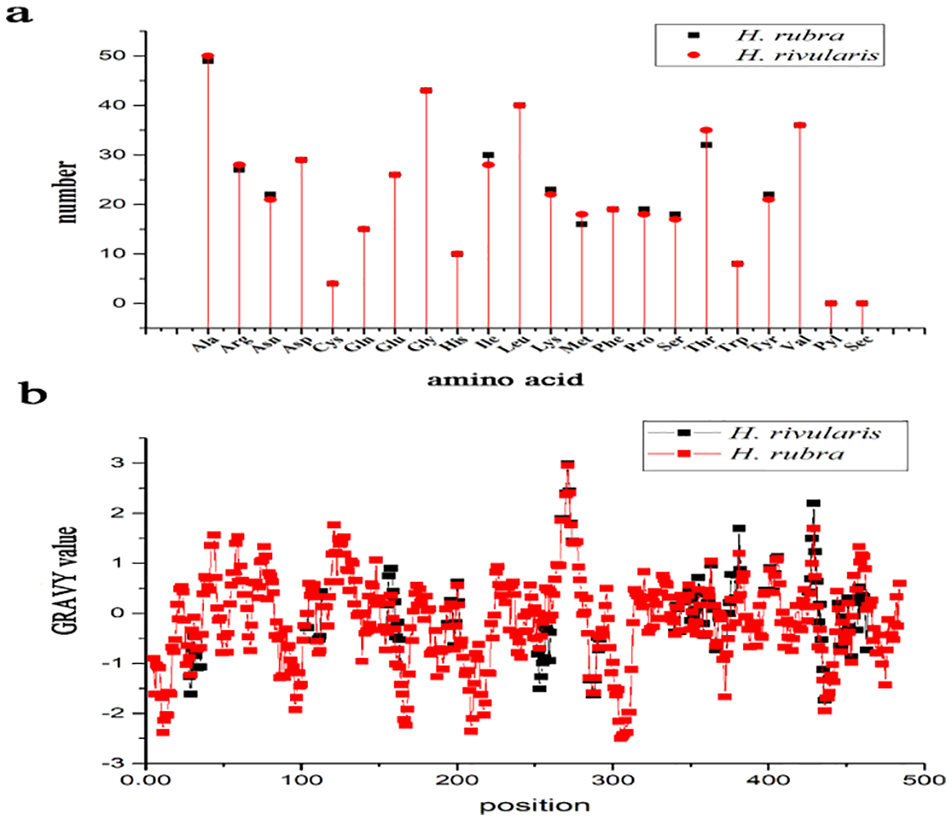
Amino acid composition (a) and hydrophobicity/hydrophilicity profile (b) of rbcL protein of Hidenbrandia rivularis and H. rubra.
There are some differences in protein secondary structure predicted by Garnier-Robson and Chou-Fasman method. The Garnier-Robson method predicted 25 α-helixes, 37 β-sheets, 22 turns, and 19 random coils of the Rubisco large subunit of H. rivularis and 32 α-helixes, 39 β-sheets, 21 turns, and 18 random coils for H. rubra. The Chou-Fasman method predicted 20 α-helixes, 19 β-sheets, and 26 turns for H. rivularis and 19 α-helixes, 17 β-sheets, and 25 turns for H. rubra. For H. rivularis, the α-helixes predicted by the 2 methods were located at positions 3-12, 55-62, 79-86, 130-143, 248-263, 316-329, 368-375, and 425-436. β-sheets were located at positions 41-47, 69-76, 99-105, 121-129, 148-153, 268-277, 342-361, 465-470, and 478-483. The turns were located at positions 16-19, 186-188, and 308-312. The random coils predicted by the Garnier-Robson method were located at 65-68, 94-98, 154-157, and 408-413. For H. rubra, the α-helixes predicted by the 2 methods were located at positions 3-12, 55-62, 79-86, 106-115, 130-143, 248-263, 316-329, and 425-436. β-sheets were located at positions 43-47, 69-76, 99-105, 121-129, 148-153, 243-248, 268-277, 344-351, and 478-483. The turns were located at positions 16-19, 186-188, and 308-312. The random coils predicted by the Garnier-Robson method were located at 65-68, 94-98, 154-158, 383-386, and 408-413 (Figure 2).

Secondary structure prediction of Rubisco of Hidenbrandia rivularis and H. rubra: (a) 241N, 180K; (b) 241N, 250Q, and (c) 254Q, 261D.
The Rubisco large subunit of H. rivularis included 33 phosphorylation sites. Among them, 13 phosphorylation sites of serine (Ser) were located at sites 13, 18, 65, 66, 97, 116, 185, 212, 282, 302, 305, 373, and 474, respectively. There were 10 threonines (Thr) phosphorylation sites located at positions 4, 47, 69, 177, 250, 303, 333, 351, 475, and 483, respectively. Ten tyrosine (Tyr) phosphorylation sites are located at positions 16, 28, 33, 84, 101, 104, 243, 255, 440, and 472, respectively (Figure 3a). The Rubisco large subunit of H. rubra included 36 phosphorylation sites. Among them, 14 phosphorylation sites of serine (Ser) were located at sites 13, 18, 65, 66, 97, 116, 185, 212, 282, 302, 305, 373, 429, and 474, respectively. There were 12 threonines (Thr) phosphorylation sites located at positions 4, 47, 69, 159, 177, 303, 333, 351, 355, 378, 475, and 483, respectively. Ten tyrosine (Tyr) phosphorylation sites are located at positions 16, 28, 33, 84, 101, 104, 243, 255, 440, and 472, respectively (Figure 3b).

Phosphorylation site prediction of Rubisco of Hidenbrandia rivularis (a) and H. rubra (b).
Adaptive evolution analysis
The results of adaptive evolution analysis were presented in Supplemental Tables S3 and S4. In the branch model, the free-ratio model revealed that the ω values of clade A-E were all less than 1, indicating that each clade is under negative selection pressure. In the 2-ratio model, 5 clades A-E were selected as foreground clades. Similarly, the ω-values of the foreground clades were all less than 1. The LRT test on 1-ratio, free-ratio and 2-ratios revealed the results of clade A (P = .003) and clade E (P = .027) were credible.
In the site model, M3 model was significantly better than M0 (P < .01), showing that the selective pressure of each site was different. The ω value of M2a was 1 and M8 (beta & ω) was larger than 1 but no positive selection sites were detected. The LRT test refused the hypothesis of positive selection sites exists in the rbcL gene.
In the branch-site model, 2 positive selection sites were detected in clade A: 124V and 181N. Six positive selection sites were detected in clade C: 254Q, 260Q, 261D, 264S, 268V, and 278A. Two positive selection sites were detected in clade D: 25L and 248L. Four positive selection sites were detected in clade E: 260Q, 264S, 268V, and 278A. LRT test was carried out for alternative hypothesis model and the corresponding null hypothesis model of each clade, and there was no credible results.
Coevolutionary analysis
rbcL gene sequence of genus Hildenbrandia was compared with the resolved 3-dimensional structure model (PDB ID:1BMV) to determine the relative position of the amino acids site. A total of 41 coevolutionary groups were calculated based on the correlation values of the amino acid pairs (Supplemental Table S5). The pairs of amino acid sites coevolving within each group were detected for the compensatory mutations based on calculation of correlation in the evolution of the hydrophobic or molecular weight characteristics of 2 amino acid sites in a multiple sequence alignment. Results of compensatory mutations through the analysis of correlation in the molecular weight, hydrophobicity or both of the amino acid sites detected as coevolving were listed in Supplemental Tables S6 to S8. Thirteen coevolutionary groups (45 pairs) based on amino acid hydrophobicity correlation, 13 groups (39 pairs) based on the correlation value of amino acid molecular weight and 11 groups (35 pairs) based on both molecular weights and hydrophobicities were estimated (Supplemental Tables S6-S8). The coevolving sites with high correlation coefficients were located in the 3-dimensional structure as Figure 4.

Location of 3 pairs of coevolution between amino acid sites of Hidenbrandia rivularis (a) and H. rubra (b).
Discussion
Amino acid compositions of Rubisco large subunit of marine and freshwater Hildenbrandia were similar with a preponderance of Ala and Gly and low proportion of Cys and Trp, which was in consistent with marine red alga Pyropia yezoensis. 35 This physicochemical characteristic is similar to the late embryogenesis abundant (LEA) proteins, which play a role in protection against heat stress, indicating the Rubisco protein of genus Hildenbrandia was heat stable. 36 The hydrophobicity of a protein is closely related to its transmembranal property. 37 The results of the physico-chemical properties showed that the Rubisco large subunit of Hildenbrandia is hydrophilic. There was slight difference in the GRAVY values of marine and freshwater Hildenbrandia species. The GRAVY values of genus Hildenbrandia were higher compared with GRAVY values of marine red alga previously reported (−1.086 for Pyropia yezoensis protein) and lower than the freshwater red alga Compsopogon (−0.099).35,38 GRAVY values are grand average hydropathy values, calculated according to Kyte and Doolittle and increasing GRAVY values indicate increasing hydrophobicity. 39 Therefore, it is inferred that the hydrophilicity of Rubisco large subunit of Hildenbrandia was higher than Compsopogon and lower than Pyropia yezoensis. The secondary structure of Rubisco large subunit of Hildenbrandia was primarily composed of α-helixes and β-sheets, revealing the relatively stable structure of this protein. The protein phosphorylation is common in post-translational modifications of proteins and is considered as a molecular mechanism for adaption evolution. 40 The determination of protein phosphorylation sites is an essential part of studying the function of target genes and their encoded products. The prediction server on the Internet at http://www.cbs.dtu.dk/services/NetPhos/ has been shown to be sufficiently accurate. 41 A total of 33 and 36 phosphorylation sites of the protein encoded by the rbcL gene in Hildenbrandia were predicted, suggesting post-translational modifications of this protein is common. The numbers of predicted phosphorylation sites in Rubisco large subunit of freshwater H. rivularis and marine H. rubra were different. More freshwater and marine species of genus Hildenbrandia should be studied in further investigation to test whether this trait is related with salinity or it correspond to variability among species.
Rubisco, the most abundant enzyme on the Earth and responsible for all photosynthetic carbon fixation, is often thought of as a highly conserved enzyme. Analysis based on phylogenies constructed from rbcL sequences of red and Chromista algal groups showed none of the algal groups displaying a signal of positive selection. It was inferred that positive selection in rbcL was prior to the divergence of large algal taxonomic groups, but not within the groups. 42 Similarly, there is no evidence of adaptive evolution in genus Hildenbrandia in our study, indicating that the rbcL gene in genus Hildenbrandia evolved either neutrally or under purifying selection. According to current reports on adaptive evolution of freshwater red algae, most groups showed no signal of adaptive evolution and were under strong negative selection pressure,38,43 except for the identification of 3 positive selection sites in rbcL gene of Bangia atropurpurea, Sheathia arcuata and Batrachospermum helminthosum. 16 On the other hand, positive selection of rbcL was detected in hot spring cyanobacteria and it is quite common in terrestrial land plants.8,44 It was speculated that the rare positive selection in cyanobacteria, algae and aquatic land plants may possibly be explained by more stable conditions of aquatic environment compared to terrestrial one. 8
Coevolution of amino acid residues is one of the key forces shaping proteins. The main reason for the amino acid sites interdependence is that proteins’ functions rely on their 3-dimensional structure and the complex functional and structural interaction networks. Functionally related amino acid residues are tightly evolutionarily linked because mutations at one position may very likely have dramatic effects on the dependent amino acid positions. 45 The coevolutionary groups among amino acids sites in protein encoded by rbcL in Hildenbrandia were more in number and the spatial positions of these sites in 3-dimensional structures of the protein were more closely related than previously reported red algae. 16 Coevolution of residues has proved common in Rubisco of land plants and coevolving sites are in closer proximity in the tertiary structure of the Rubisco large subunit. 46 This phenomenon was also found in the freshwater red algal genus Hildenbrandia. Hildenbrandia is an anciently derived lineage and its basal position among the members of Florideophyceae has been acknowledgmented. 47 Despite the long evolutionary history, rbcL gene in genus Hildenbrandia under different environments is rather conservative.
Supplemental Material
sj-pdf-1-evb-10.1177_1176934320977862 – Supplemental material for Analysis of Adaptive Evolution and Coevolution of rbcL Gene in the Genus Hildenbrandia (Rhodophyta)
Supplemental material, sj-pdf-1-evb-10.1177_1176934320977862 for Analysis of Adaptive Evolution and Coevolution of rbcL Gene in the Genus Hildenbrandia (Rhodophyta) by Nan Fangru, Han Yuxin, Liu Xudong, Feng Jia, Lv Junping, Liu Qi and Xie Shulian in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are grateful to the National Natural Science Foundation of China (No. 31670208, 41871037 to SX and 31800172 to FN) and the Fund for Shanxi “1331 Project” for funding this project.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
NF performed the analysis and wrote the paper. HY, LX, FJ, LVJ, and LQ gave technological help. XS designed the experiments and modified the manuscript. All authors read, commented, and approved the final paper.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.