
Abstract
Dengue is an infectious disease caused by dengue virus (DENV) and transmitted between human hosts by mosquitoes. Recently, Indonesia was listed as a country with the highest cases of dengue by the Association of Southeast Asian Nations. The current treatment for dengue disease is supportive therapy; there is no antiviral drug available in the market against dengue. Therefore, a research on antiviral drug against dengue is very important, especially to prevent outbreak explosion. In this research, the development of dengue antiviral is performed through the inhibition of n-octyl-β-D-glucoside (β-OG) binding pocket on envelope protein of DENV by using analogs of β-OG pocket binder. There are 828 compounds used in this study, and all of them were screened based on the analysis of molecular docking, pharmacological character prediction of the compounds, and molecular dynamics simulation. The result of these analyses revealed that the compound that can be used as an antiviral candidate against DENV is 5-(3,4-dichlorophenyl)-N-[2-(p-tolyl) benzotriazol-5-yl]furan-2-carboxamide.
Introduction
During the last few decades, dengue fever has been the most common infectious disease in more than 100 tropical and subtropical countries. It threatened >2.5 billion human life.1–3 Every year, there are ~100 million cases of dengue fever. Among them, 500,000 cases lead to hospitalization and 25,000 cases lead to death. 4 According to the data from all over the world, Asia is ranked as top one in terms of dengue cases per year. Meanwhile, according to World Health Organization, Indonesia has the highest number of dengue infections in Southeast Asia. 5 Dengue has become a public health problem during the last 41 years in Indonesia. In 1968, dengue was found for the first time in Surabaya, and since then the number of cases has continued to rise, from 58 cases in 1968 to 158,912 cases in 2009. There is also a massive increase in the number of endemic provinces and cities, from 2 provinces and 2 cities to 32 (97%) provinces and 382 (77%) cities, respectively, in 2009. 5
Despite a huge number of dengue infections all over the world yearly, there is no effective therapeutic treatment available in the market until now. The current method for controlling the spread of the disease is by controlling its vector, Aedes aegypti. The currently available healing methods are supportive therapies that involve body fluid replacement therapy, analgesic administration, and total bed rest. Scientists have also tried to develop vaccine to reduce the incidence rate of dengue fever; however, the world's first dengue vaccine is still undergoing phase 3 clinical trial and is expected to be in the market by 2016. This vaccine only gives protection from three out of five serotypes of dengue virus (DENV). 6 The existence of these five serotypes of DENV has hindered the efforts of developing effective vaccine against them. Infection by one DENV serotype does not give protective immunity against other serotypes. In fact, subsequent infection by a different DENV serotype has led to the increase in viral replication and the escalation of disease severity into dengue hemorrhagic fever and dengue shock syndrome through a process known as antibody-dependent enhancement.7,8 Hence, an effective vaccine must give protective immunity against all five serotypes of DENV at once. Based on these reasons, the development of antiviral agent as a therapeutic option is urgently needed, especially in times of outbreak. Over the past few decades, major advances have occurred in the development of antiviral drugs against DENV. Several approaches have been reported, including inhibitor of nucleoside triphosphate, 9 suppressor of viral RNA synthesis, 10 inhibitor of protease and helicase activities of DENV NS3, 11 peptides that mimic the conserved protein cleavage sites, 12 host alpha-glucosidase inhibitors that suppress viral secretion and infection,13,14 inhibitor of c-Src protein kinase that inhibits viral assembly and maturation, 15 monoclonal antibody,16,17 inhibitor of domain III of fusion protein, 18 inhibitor of envelope protein, 19 and polyanions preventing viral binding to host cell receptor. 20
In silico screening of database of chemical compounds has currently evolved as a promising approach to identify lead compounds. In silico approach has become inseparable part of drug design and development. Although in silico screening requires the knowledge of three-dimensional (3D) structure of the target, it has generated satisfying result in a number of systems.21–24
Previous research revealed that the crystal structure of the soluble ectodomain of DENV-2 envelope has a hydrophobic pocket residing in the hinge region between domains I and II. This hydrophobic pocket binds to a small detergent molecule, n-octyl-β-D-glucoside (β-OG); therefore, it is known as β-OG binding pocket and was proposed as an appropriate target for developing small-molecule inhibitors of viral-host fusion process. 25
In this research, we searched antiviral candidates through virtual screening of chemical compounds using β-OG pocket as the target protein. The compounds used in this research are commercially available analogs (90% resemblance) of β-OG pocket binder compounds (β-OG, as a natural ligand of this pocket) and the compounds used in Poh et al's, 22 Kampmann et al's, 23 and Yennamalli et al's research. 21
This research aims to find new antiviral candidates against DENV that are analogs of β-OG pocket binder of DENV envelope protein according to previous research21–23,25 through molecular docking and dynamics simulation. This research is expected to serve benefit in the efforts of antiviral development against DENV and to be available in the market; hence its existence would help to reduce the incidence rate of dengue fever.
Research Methodology
Tools and materials
This research was conducted in silico by using bioinformatics tools.26,27 Several online and offline software were used in this research. Offline software used in this research were Molecular Operating Environment (MOE) 2008.10, ACD/Labs' ChemSketch 12.01, Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) Ligand Explorer 4.1.0, VEGA ZZ 3.0.5, and Toxtree 2.5.0. The materials used in this research are sequence data and 3D structure of envelope protein of DENV. These data are available online on National Center of Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov), EMBL-EBI (http://www.ebi.ac.uk/) and PDB at the RCSB website (http://www.rcsb.org/pdb/home/home.do). The analogs of β-OG pocket binder are obtained from ZINC database (http://zinc.docking.org), ChemSpider (http://www.chemspider.com), and PubChem (http://pubchem.ncbi.nlm.nih.gov/) websites.
Procedure
Searching of 3D structure of DENV envelope protein
Searching the right structure of DENV envelope protein to be used in the drug design is one of the most important things to make sure that the selected structure represents other related envelope proteins. The 3D structure of DENV envelope protein could be looked up at PDB of RCSB. The selected structure of this research is 1OAN, which is a crystal structure of the DENV-2 envelope protein. This structure was determined primarily by Modis et al. 25 They reported a ligand-binding pocket in the structure of DENV envelope. The 3D structure of DENV envelope was then saved in the PDB format. This structure was then used in molecular docking and molecular dynamics process.
Sequence similarity searching of DENV envelope protein
Sequence similarity searching through sequence alignment is required to make sure that the selected sequence of DENV envelope protein represents other related sequence and to seek conserved residue. The tools used in the alignment were NCBI Basic Local Alignment Search Tool (BLAST). The envelope protein sequence of 1OAN was uploaded to the NCBI BLAST website, and Blastp program was selected. A total of 10 related sequences of DENV envelope proteins from each serotypes were compared. The total number of representative sequences was 40. These sequences were compared with each other using the option “align to or more sequences” in Blastp.
Ligand selection
The ligands used in this research were analogs of compounds that showed good interaction with β-OG pocket of DENV envelope. Based on previous research, there are several compounds that are able to bind to β-OG pocket. These compounds are β-octyl glucoside from Modis et al's research; 25 A1, A2, A3, A4, and A5 from Kampmann et al's research; 23 NITD448 from Poh et al's research; 22 and R1, R2, R3, R4, R5, R6, and R7 generated from Yennamalli et al's research. 21 The commercially available analogs of these compounds were then searched on ZINC database, PubChem, and ChemSpider websites. The ligands were drawn using ChemSketch and saved in .mol format.
Molecular docking preparation
The targeted protein and the ligands must be prepared before conducting molecular docking process. The targeted protein, in this case, representative envelope protein of DENV, was opened using MOE. The undesirable amino acid side chain, the attached ligand/inhibitor, and the water molecules of the protein were removed from the protein sequence. The protein was protonated to add polar hydrogen to the structure, since its crystal structure that was obtained from X-ray crystallography did not contain hydrogen atom. This step was conducted using compute > protonate 3D menu in MOE. The partial charge was then applied to the protein by hitting partial charge menu in MOE. Energy was minimized using energy minimize menu in MOE to obtain protein conformation with the lowest energy. The selected force field was AMBER99, which was parameterized for proteins and nucleic acids, while the chosen solvation was the gas phase.
Analogs of β-OG binder as the ligands were obtained from ZINC database in .mol2 format; the structures were then converted into 3D format using VEGA ZZ and then opened on database viewer of MOE. The ligands went through several steps of preparation, including wash, partial charge, and energy minimization. The selected force field in these processes was Merck Molecular Force Field 1994 since it is parameterized for small organic molecules in the gas phase. After preparation process, protein and ligands were ready for molecular docking simulation.
Molecular docking simulation
The pipeline for molecular docking and dynamics simulation followed established pipeline from previous research.28,29 Molecular docking was conducted using menu compute > simulation > dock in MOE 2008.10. The selection of active site residues of envelope protein is crucial before docking process and was conducted by using sequence editor in MOE. The number of docking pose to capture is 100 with only 1 best pose showing the final result. The interaction between DENV envelope protein and ligands was visualized using LigX and surface and maps menus.
Screening drug candidate
The ligands as drug candidates were screened according to their absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. This screening was performed using several software, including ACD/I-Lab, Toxtree v2.5.0, FAF-Drugs 2, and Molinspiration. Several parameters were observed, including their physicochemical characters according to Lipinski's rule of five (RO5), 30 oral bioavailability, mutagenicity and carcinogenicity, and health effect probabilities.
Molecular dynamics simulation
Molecular dynamics simulation was performed using MOE 2008.10. 31 The selected solvation mode is generalized Born implicit solvent, while the force field used is AMBER. This simulation consists of three steps: initialization, equilibration, and production.32,33 The initialization process was conducted for 100 picoseconds at 300 K. The time needed to conduct equilibration process was determined according to initialization time when a ligand starts to form a stable complex with the DENV envelope. The production step took 10,000 picoseconds to run and involved a cooling stage for 10 picoseconds.
Results
Searching of 3D structure of DENV envelope protein
The structure of 1OAN consists of two chains, A and B; each of them consists of 394 amino acids. The structure also comprises several ligands component: beta-D-mannose (BMA), alpha-L-fucose (FUC), sodium ion (NA) and N-acetyl-D-glucosamine (NAG), but their positions are not close to β-OG binding pocket based on the analysis of RCSB PDB Ligand Explorer 4.1.0 (Fig. 1). NAGs are located near residue 67 of chains A and B, residue 159 of chain A, residue 149 of chain B near FUC and BMA, and residue 157 of chain A where FUC and BMA are attached to it.
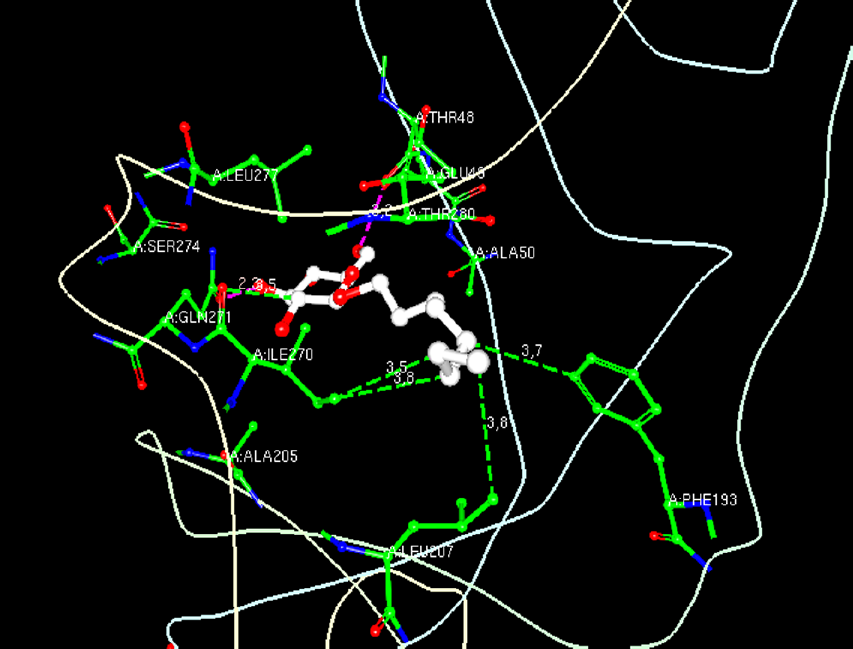
The contact residues of DENV-2 E protein in complex with β-OG.
Sequence similarity searching
The result of sequence similarity searching using NCBI BLAST toward the sequences of all types of DENV revealed that the sequence of 1OAN is almost identical with other DENV-2 sequences found in the NCBI website. Their identity score is 97%. However, the identity of 1OAN sequence and other type of DENV sequences fall between 64% and 69%, with the least identity obtained when 1OAN was compared with the envelope protein of DENV-4. The higher the identity of two or more sequences, the more similar their protein structure. If the target proteins share the identity >50%, their protein structure is sufficiently reliable for drug design purpose. 34
Ligand selection
The structure of β-octyl glucoside used in Modis et al's group; 25 A1, A2, A3, A4 and A5 from Kampmann et al's research; 23 NITD448 from Poh et al's research; 22 and R1, R2, R3, R4, R5, R6, and R7 generated from Yennamalli et al's 21 research were drawn on ZINC database, ChemSpider, and PubChem. Then the analogs with 90% identity were searched using query tools. The results were drawn using ChemSketch. The total number of ligands used in this research were 828 ligands: 395 compounds were analogs of β-OG, 3 compounds analogs of A1, 1 compound analog to A2, 2 compounds analogs of A3, 1 compound analog of A4, 2 compounds analogs of A5, 1 compound analog of NITD448, 131 compounds analogs of R1, 60 compounds analogs of R2, 13 compounds analogs of R3, 65 compounds analogs of R4, 36 compounds analogs of R5, 1 compound analog of R6, and 117 compounds analogs of R7.
Molecular docking
Molecular docking was conducted to search the most stable bonding conformation between the ligand and the target protein. This process was conducted twice toward 828 ligands and 14 standards (β-OG, A1, A2, A3, A4, A5, NITD448, R1, R2, R3, R4, R5, R6, and R7) against binding site residues of β-OG pocket (Thr 48, Glu 49, Ala 50, Phe 193, Ala 205, Leu 207, Ser 274, Gln 271, Leu 277, and Thr 280). 25 These contact residues were known through analysis of DENV-2 E protein in complex with β-OG using RCSB PDB Ligand Explorer 4.1.0 (Fig. 1).
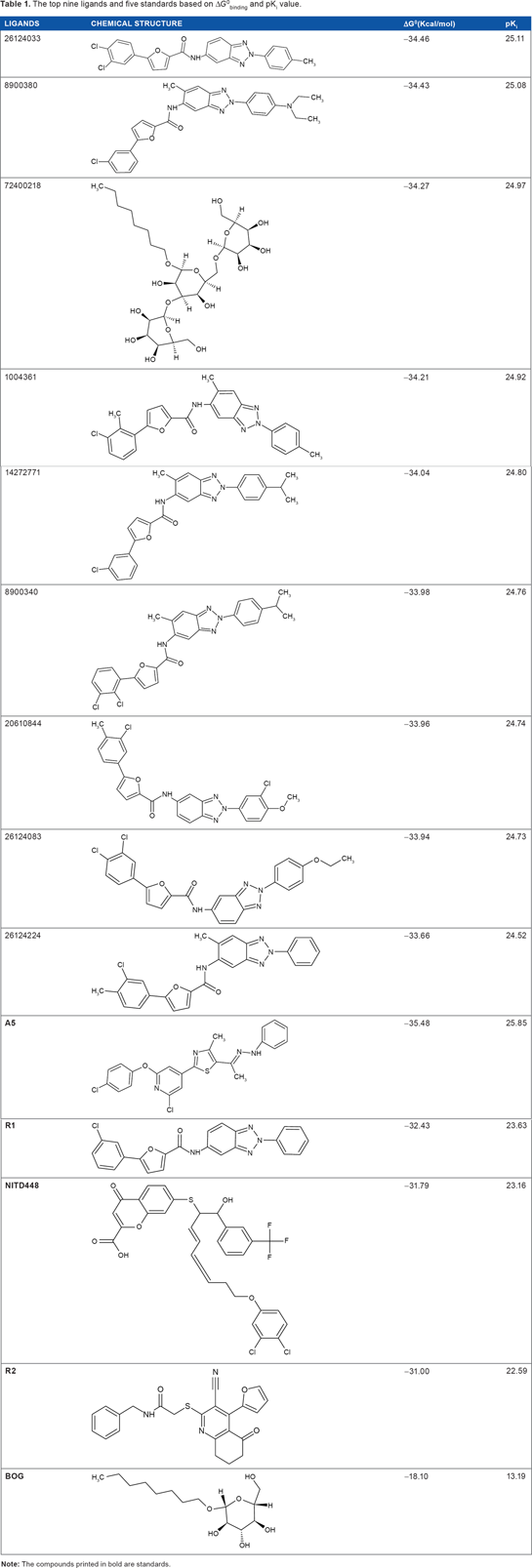
The result of molecular docking revealed nine top ligands with comparable value of ΔG0binding with standards (Table 1). The more negative ΔG0binding, the stronger the interaction between the ligand and the target protein. While the higher the pKi, the stronger the affinity of the receptor-ligand complex. In this case, ligand 26124033, which is an analog of R1, has the strongest affinity toward the β-OG pocket among all ligands. According to the value of ΔG0binding and pKi, ligand 26124033 has stronger affinity than R1 to the β-OG pocket. The structure of 26124033 and R1 is shown in Figure 2A and B, respectively.
The top nine ligands and five standards based on ΔG0binding and pKi value.
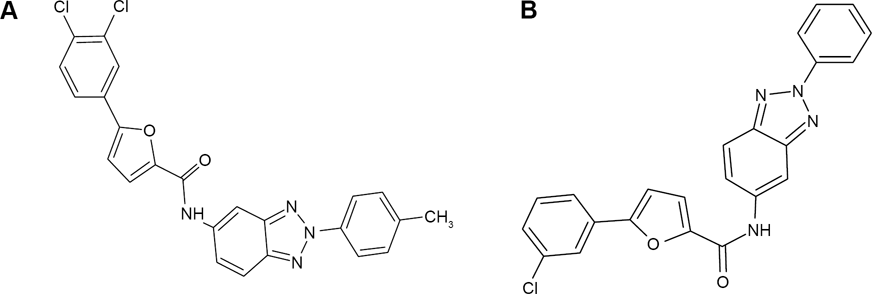
Comparison of structure between
The best five standards according to molecular docking result are A5, β-OG, NITD448, R1, and R2 (Table 1). A5 generated the most negative ΔG0binding of all standards, which was also more negative than the best ligands displayed in Table 1, 26124033. There were several compounds generated from molecular docking that possessed far lower ΔG0binding than A5. However, all of them have shown poor pharmacological characteristics upon prediction. Therefore, the results displayed in Table 1 representing the ligands with slightly lower value than or comparable value of ΔG0binding with standards yet possess good pharmacological properties.
The structure of 26124033 and R1 differ from the structure of 26124033 by an extra methyl and chlorine group (Fig. 2). This difference leads to a distinct value of ΔG0binding and the interaction of ligand-receptor. Ligand 26124033 formed hydrogen bond with the side chain of Gln 200 and the backbone of Trp 206 in the β-OG pocket, while R1 maintained hydrophobic interaction between its aromatic group and Phe 279's aromatic group of target receptor (Fig. 3). In Yennamalli et al's research, 21 R1 and R2 were used to target site I of DENV envelope protein. The research generated R1 as the best ligand to target that site and suggested that R1 might inhibit different targets of DENV protein. Based on molecular docking result in our research, it is revealed that R1 also has strong interaction with the β-OG binding pocket of DENV envelope protein. However, its standard binding free energy with the target receptor is higher than its analog and A5.

The interaction scheme between β-OG pocket and (A) 26124033 and (B) R1.
A5 is the best ligand generated from Kampmann et al's research 23 that targeted β-OG pocket. Our research confirms that A5 does indeed have strong affinity with the target receptor as represented by its lowest ΔG0binding value among top ligands and standards. A5 maintained strong hydrophobic interaction with Phe279 of β-OG pocket (Fig. 4).
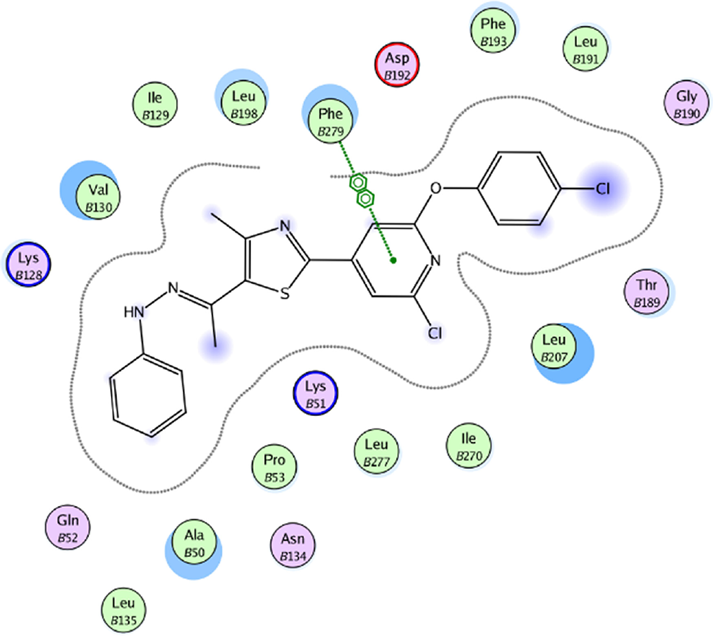
The interaction scheme between β-OG pocket and A5.
Analysis of pharmacological prediction
The pharmacological characteristic of drug candidates was predicted using several software, including ACD/I-Lab, FAF-Drugs 2, and OSIRIS Property Explorer. Lipinski's RO5 is a good approach for designing a drug that will be administered orally. Most drugs that pass clinical trials fail to reach the market because they are not orally active. Therefore, a drug candidate that conforms to these five rules will have increased probability to reach the market. An orally active drug should not violate more than one of these criteria: (i) it should have less than five hydrogen bond donors, (ii) it should have less than 10 hydrogen bond acceptors, (iii) its molecular mass should not be more than 500 Da and (iv) its log P (octanol-water partition coefficient) value should be <5. 30 These rules involve numbers that are multiples of five; therefore, Lipinski's rule is also known as the rule of five (RO5). The results of Lipinski's rule calculation for ligands and standards are shown in Table 2.
Calculated pharmacological characteristics for ligands and standards based on Lipinski RO5.
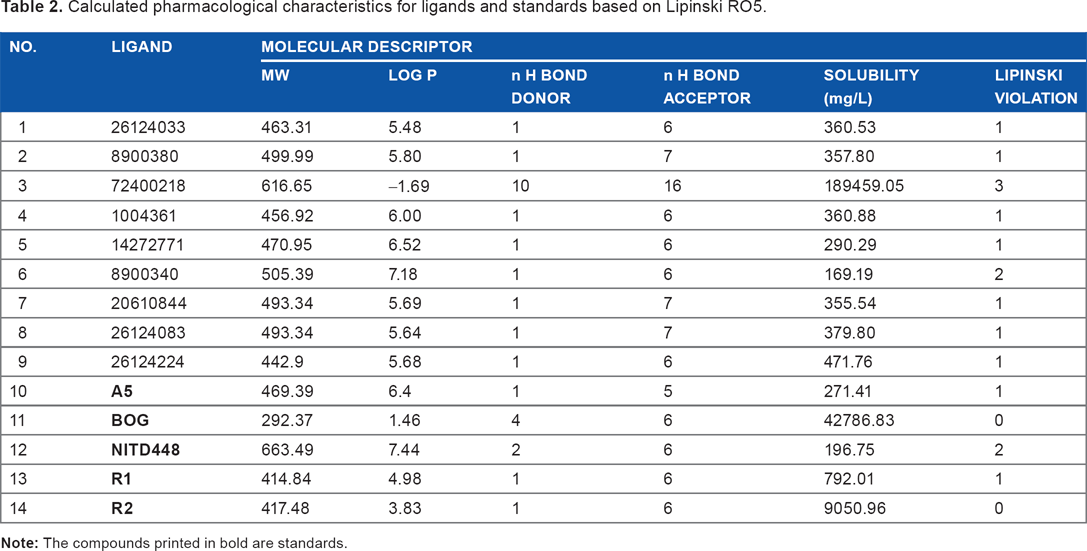
According to the data, only two ligands violate more than one rule of RO5, while the rest of them only violate one rule. RO5 is more like a guidance than an absolute rule; therefore, if a compound violates one out of five rules, the compound is still probably orally bioavailable and can be absorbed by the body. Hence, there is a possibility that all ligands, except 72400218 and 8900340, can still be easily absorbed by the body.
Among the standards, only β-OG and R2 follow RO5, while ligand R1 has the same number of violation as its analog, 26124033. Utilizing β-OG as standard did not imply that it is a drug candidate. We examine the binding properties of β-OG in order to comprehend the reactivity of the other compounds. A5 from Kampmann et al's research 23 also violates one rule of RO5, while NITD448 violates two rules due to its high molecular weight and log P. Log P represents hydrophilic character of a compound. The presence of a large number of hydrogen bond donor group tends to disrupt the compound's permeability across lipid bilayer of membrane. This can be measured indirectly by the value of partition coefficient between octanol and water (log P), since water is a highly hydrogen bonding solvent and octanol acts as a non-hydrogen bond accepting solvent. The RO5 restricts that the log P value of a compound should be <5, in order to ensure its absorption and permeation by the body.
Besides the analysis of ligand's conformity with the Lipinski's RO5, the drug likeness and drug score of ligands and standards were also calculated using OSIRIS Property Explorer. These calculations are based on several properties of the compound, including mutagenicity, tumorigenicity, irritant, reproductive effects, log P, and solubility of the compound.
There are five ligands that had no mutagenicity, tumorigenicity, irritant, and reproductive effects on health (26124033, 72400218, 1004361, 20610844, and 26124224), and therefore, they generated high drug score. Among all, β-OG generated the highest score (0.47), which indicates its highly drug-conform behavior. R1 also produced high drug score, which is higher than its analog 26124033. However, A5 failed to show harmful effect on health as it is tumorigenic according to OSIRIS Property Explorer's prediction (Table 3).
The calculated pharmacological characteristics for ligands and standards using Osiris Property Explorer.
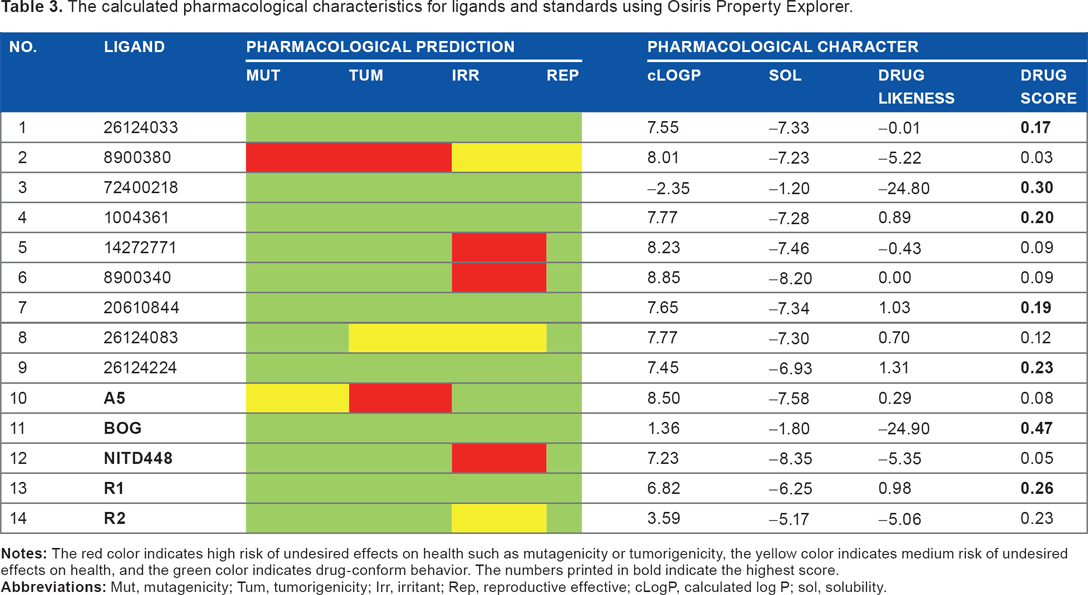
Oral bioavailability of a compound can also be predicted by considering Veber's rule. Veber observed that the compounds that meet only two criteria of RO5 (that is, having less than 10 rotatable bonds and are less than or equal to 140 å 2 ) will likely have good oral bioavailability in rats. 35
The result of oral bioavailability calculation according to Veber's rule is displayed in Table 4. It showed that all tested compounds possess good oral bioavailability, except ligand 72400218 as it breaks two rules of Veber.
Prediction of oral bioavailability of the compounds according to Veber's rule.
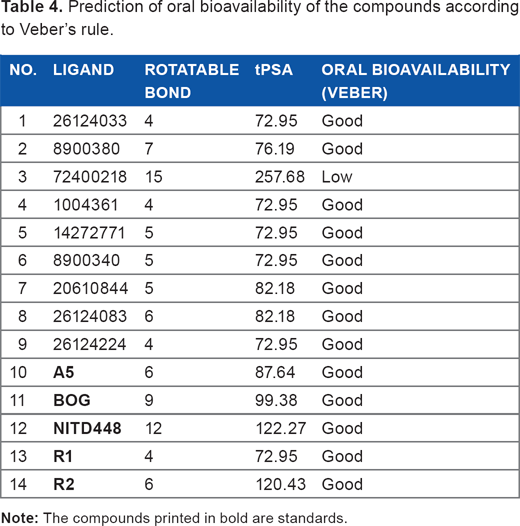
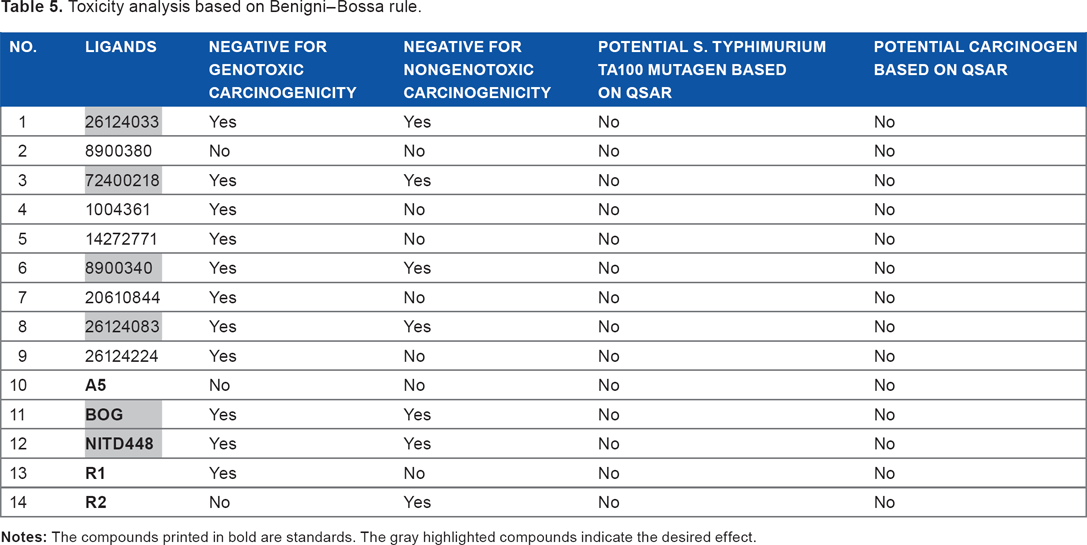
The last pharmacological prediction of the compounds was toxicity prediction using Toxtree according to Benigni-Bossa rule. This rule suggests several mutagenic and or carcinogenic functional groups, such as acyl halide, benzyl ester, epoxide, aliphatic halogen, alkyl nitrite, quinone, hydrazine, polycyclic aromatic hydrocarbon, thiocarbamate, aromatic amine, and hydroxylamine. 36 According to this rule, only four ligands and two standards had no carcinogenic and mutagenic effects on health. They are 26124033, 72400218, 8900340, 26124083, β-OG, and NITD448 (Table 5). The summary of all pharmacological prediction revealed 26124033 as the best ligand, which can be further processed for molecular dynamics simulation.
Toxicity analysis based on Benigni-Bossa rule.
Molecular dynamics analysis
The best ligand (26124033) obtained from molecular docking was analyzed based on its molecular dynamics simulation. This simulation is performed at temperature 310 K and 312 K. The conformations of the complex between the target protein (1OAN) and the ligand (26124033) during molecular dynamics simulation are captured in Figures 5–8. The temperature of 310 K was selected as the normal body temperature, while the temperature 312 K represented human body temperature during fever.

The visualization of complex conformation during initialization, equilibration, and production at 310 K.

The interaction scheme of the complex during initialization, equilibration, and production steps at 310 K.

The visualization of complex conformation during initialization, equilibration, and production at 312 K.
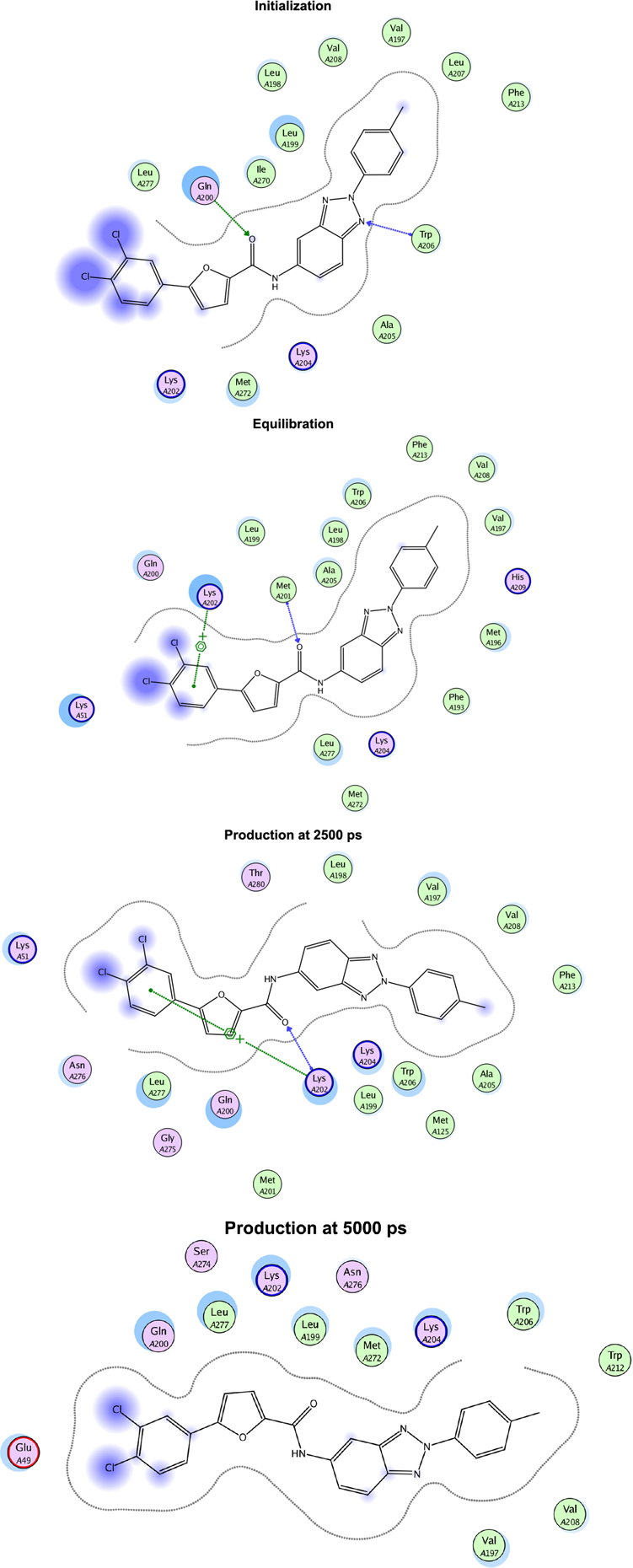
The interaction scheme of the complex during initialization, equilibration, and production steps at 312 K.
The initialization process was conducted for 200 picoseconds, which was needed by the protein-ligand complex to start interaction with the solvent and adjust its conformation. The initialization time for this complex was 160 picoseconds. During initialization, the ligand formed hydrogen bond with Gln 200 and Trp 206 of the receptor (Figs. 5 and 6).
The equilibration step involves the heating stage from 300 K to 310 K during 10 picoseconds. After heating, equilibration took place. The time needed to conduct the equilibration step was determined previously from the initialization time, which was 160 picoseconds. During this process, the structure of receptor-ligand complex changed significantly, which was marked by the fluctuation of conformational energy of the complex. The rise of temperature induced the increase of kinetic energy of each atom in the complex and solvent, which in turn led to continuous conformational change during simulation. The equilibration step aimed to ensure that the receptor-ligand complex has adjusted its conformation with the normal body condition. During equilibration step at 310 K, the ligand formed hydrogen bond with the side chain of Lys 128, while at 312 K, the ligand interacted with the receptor through hydrogen bond with Met 201 and aromaticcation interaction with Lys 202. After the stable complex conformation was achieved, simulation was then continued to the production and cooling stage.
The production step was performed to simulate complex stability in the presence of solvent during 5,000 picoseconds. A drug candidate must show good complex stability with the target protein. The complex conformations during the production stage were displayed at 2,500 picoseconds and 5,000 picoseconds. According to the interaction scheme of the complex, the ligand was well buried in the β-OG pocket of the envelope protein. The stronger interaction of the complex was observed during 2,500 picoseconds than during 5,000 picoseconds at both temperatures. While at temperature 312 K, the ligand mainly formed weak interaction with the target receptor at the end of the production stage (Figs. 7 and 8).
The stability of the receptor-ligand complex was then evaluated through the plot between root-mean-square deviation (RMSD) and simulation time that was produced in the production step. The RMSD represents the magnitude of conformational change of the complex during molecular dynamics simulation. The RMSD curve showed that the RMSD value of complex at 310 K is lower than 312 K, which suggested that the conformational change of the complex occurred more frequently at 312 K than at 310 K (Fig. 9). The insertion of ligand into β-OG pocket aims to avoid the conformational change of DENV envelope protein from dimer into trimer. Therefore, it can be inferred that the ligand 26124033 could stabilize the envelope protein conformation more effectively at 310 K than at 312 K.
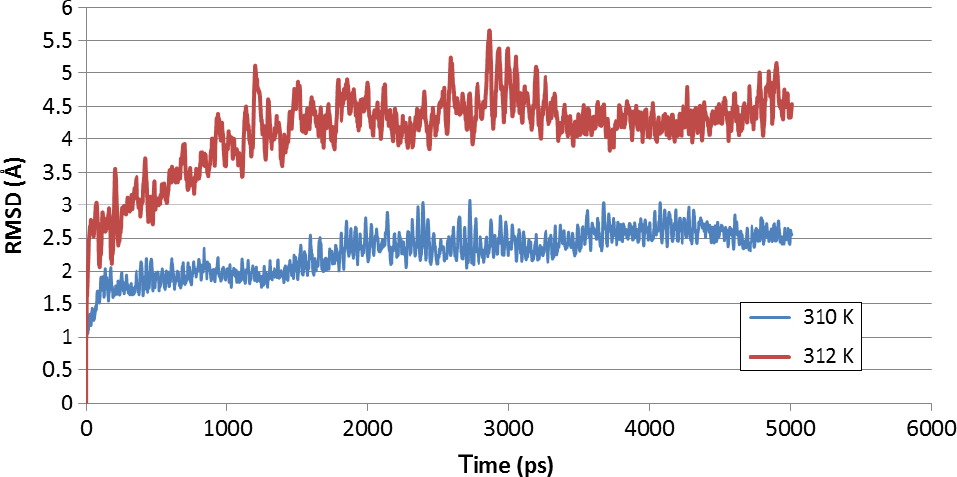
The RMSD curve of the receptor-ligand complex.
Based on molecular dynamics simulation, ligand 26124033 can maintain stable interaction with β-OG binding pocket during simulation time (5,000 picoseconds) at temperatures 310 K and 312 K. The ligand was still well buried until the end of simulation. Therefore, this ligand can be used as a lead compound for dengue antiviral drug development. The IUPAC name of this ligand is 5-(3,4-dichlorophenyl)-N -[2-(4-methylphenyl)-2H-benzotriazol-5-yl]furan-2-carboxamide, with the molecular formula C24H16Cl2N4O2.
Discussion
Our pipeline has joined several different computational methods to produce the best lead compounds from target determination, namely molecular docking, ADMET, and molecular dynamics (Fig. 10). 37 This pipeline is in accordance with the standard already in place for structure-based functional design of drugs. 38 In this respect, nowadays, computer-based programs are indispensable for efficient drug design. 39 Owing to its speed and less resource intensiveness, the structure-based drug design can test unavailable compounds as shown in our approach. 40
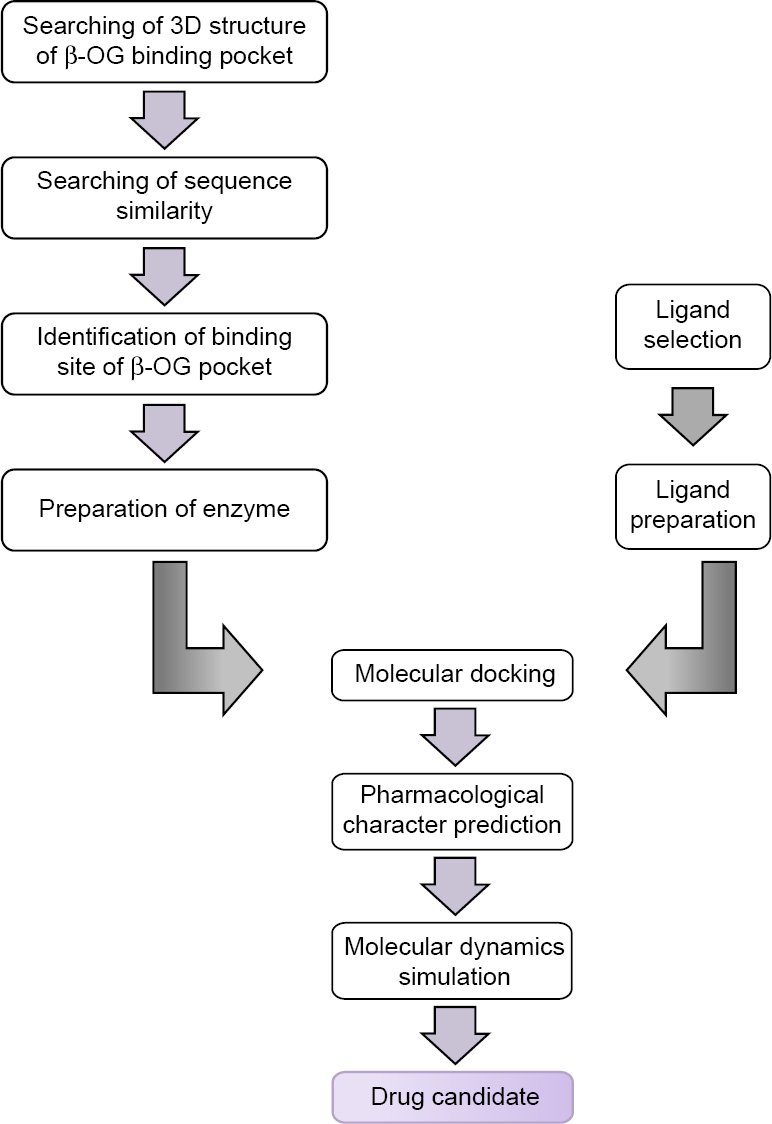
A research flowchart.
Ligand 26124033, which is an analog of R1, could be made on demand by a company listed in the ZINC database. 41 R1 could be bought from chemical companies. 21 However, how to synthesize 26124033 from R1 is already decided as it could be bought from companies as well. The availability of β-OG pocket as the binding pocket is also guaranteed for flavivirus in general, so the utilization of this pocket in DENV is feasible as well. 42
The complex stability that formed by the DENV envelope protein and 26124033 in the normal body temperature suggests that this ligand may better serve prophylaxis than treatment. The formulation of 26124033 as a prophylaxis agent would eventually make it more useful to ward of dengue infection. The existence of chlorine functional groups more abundantly in 26124033 than R1 could enact concern about the toxicity of our best compound. Although the ADMET testing has shown that 26124033 is indeed a safe compound, its reactivity in in vitro and/or in vivo assay is still unknown. However, strategy could be devised in order to ward off safety concern of this compound. Constructing prodrug is one of the options to increase the safety of 26124033.43–45 Having prodrug to deliver the lead compound is one of the safest way to utilize it in the clinical trial. Thus, if the in vitro and in vivo research proved that the compound of 26124033 is not effective, other compounds such as 72400218, 8900340, and 26124083 could be a viable option to enter wet laboratory due to the absence of carcinogenic and mutagenic effects on health.
Conclusion
Screening of β-OG pocket binder analogues was done for 828 ligands and targeting β-OG binding pocket of DENV-2 envelope protein. The study of molecular docking, pharmacological prediction, and molecular dynamics revealed that 26124033 has strong interaction with the β-OG binding pocket, has good pharmacological properties, and maintains stable conformation with the target protein. The IUPAC name of this ligand is 5-(3,4-dichlorophenyl)-N-[2-(p-tolyl) benzotriazol-5-yl]furan-2-carboxamide.
Author Contributions
USFT and AAP supervised this research. HZ worked on the technical details. SI gave technical assistance to the whole process. DK gave crucial suggestion to improve our pipeline. All authors are responsible for writing this manuscript.
Footnotes
Acknowledgment
Thanks go to Directorate of Research and Community Engagement, University of Indonesia, for providing facilities for this research.