
Abstract
Ever increasing propensity of antibiotic resistance among pathogenic bacteria raises the demand for the development of novel therapeutic agents to control this grave problem. Advances in the field of bioinformatics, genomics, and proteomics have greatly facilitated the discovery of alternative drugs by swift identification of new drug targets. In the present study, we employed comparative genomics and metabolic pathway analysis with an aim of identifying therapeutic targets in Mycoplasma hominis. Our study has revealed 40 annotated metabolic pathways, including five unique pathways of M. hominis. Our study also identified 179 essential proteins, including 59 proteins having no similarity with human proteins. Further filtering by molecular weight, subcellular localization, functional analysis, and protein network interaction, we identified 57 putative candidates for which new drugs can be developed. Druggability analysis for each of the identified targets has prioritized 16 proteins as suitable for potential drug development.
Introduction
Mycoplasmas are cell wall-deficient, smallest, free-living organisms, which are resistant to many commonly used antimicrobial agents. 1 Although most of the Mycoplasma are naturally found as commensals in the genitourinary tract, three species are well-known pathogens: Mycoplasma pneumoniae, M. Hominis, and M. Genitalium. 2 To lead a parasitic lifestyle, M. Hominis resides in the urogenital tract of women and sexually active men where it gains necessary nutrients. 3 Sometimes, the bacterium is found in a symbiotic relationship with protozoa, Trichomonas vaginalis, an event that allows for more successful transfer of M. hominis from one person to another, and increases its resistance to antibiotics.3,4 This bacterium is the causative agent of numerous health problems, such as pelvic inflammatory disease, bacterial vaginosis, postpartum fever, and infertility in females.2,5–7 In addition, M. hominis is capable of causing diseases of the central nervous system in newborn babies 8 and is associated with prostate cancer. 9
Currently, antibiotics are the only available treatment option against M. hominis infection. However, this species is inherently resistant to beta-lactam group antibiotics and macrolides because of their lack of cell wall and a mutation in 23S rRNA, respectively. 10 Moreover, M. hominis has been found to carry resistance trait against ciprofloxacin and ofloxacin. 11 The increasing trend of antibiotic resistance demands the discovery of alternative therapeutic agents for the treatment of infection caused by this bacterium. These issues require the need for exploring new drug targets in this bacterium, which will create the avenue for new drug discovery or will augment the sensitivity of the currently existing antimicrobial agents.
Increasing availability of related genomics, proteomics, metabolomics, and many other omics data of the infectious agents and information about molecules that can alter the capability of survival of the infectious organism have facilitated the search for new drugs or drug targets. As the genome of M. hominis, the second smallest genome among the free-living organisms, has already been sequenced, similar kind of study can also be possible for this bacterium. 12 Numerous in silico methods have already been developed for the prediction of potential drug targets in many bacteria and fungi.13–16 These approaches predict drug targets by comparing the proteins present in an infectious agent to those present or absent in the host. Proteins that are unique to the infectious agent and essential for its survival can be potential therapeutic targets. In principle, any essential protein present in the organism can be targeted to control the growth of that organism. 17
In the present study, we performed genomics, proteomics, and metabolic pathway analysis of M. hominis with an aim of identifying new drug targets. This study identified several drug targets and available inhibitors for those targets. We expect that present findings will not only extend our understanding of the molecular pathogenesis of M. hominis, but also open a new window to develop novel therapeutic agents against this deadly pathogen.
Methods
Identification of pathogen and host metabolic pathways
Consideration of safety is the prime issue for the development of any therapeutic agent. If the therapeutic agent is not safe for the host, it will never get regulatory approval. Although the overall similarity between eukaryotic and prokaryotic cell is very limited, the similarity in the coding region of a particular gene or functional domain of any protein may result in cross-reactivity of a therapeutic agent against the host. In addition, if a drug inhibits any essential protein of the host, it might produce severe side effects to the host species. Therefore, the starting point of this study was to compare the metabolic pathways of the pathogen and the host species. Information of metabolic pathways was extracted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database.18,19 Metabolic pathways and respective identification numbers of M. hominis and H. sapines were retrieved from the NCBI database. Subsequently, a manual comparison was made and pathways that appeared only in M. hominis but not in humans were selected as unique, and the remaining pathways of the pathogen were categorized as common pathways. Proteins were identified from the unique and common pathways and the corresponding amino acid sequences were downloaded from the NCBI database.
Screening of essential genes
Genes that are indispensable to supporting cellular life are called essential genes. Database of Essential Genes (DEG) includes all of the essential genes that are currently available. To identify essential genes of M. hominis, proteins of unique and common pathways were subjected to BLASTp against DEG with an E-value cut-off of 10-10 and with a minimum bit score of 100. 20
Identification of non-host essential proteins
After the identification of essential proteins, they were compared with the human non-redundant protein sequence database (nr) to identify non-host proteins of M. hominis. A BLASTp analysis was performed and proteins without hits below the threshold E-value of 0.005 and <35% identity were picked out as non-host essential proteins. 21
Molecular weight determination and protein 3-D structure identification
Molecular weight (MW) of each of the potential targets was determined using online tools followed by confirmation with the available literature. Previous literature suggested that smaller proteins are suitable targets for drug development because they are more soluble and easier to purify in comparison with large proteins. 22 Proteins having MW larger than 110 kDa were excluded, and the resultant list of proteins was enlisted as sigma (∑) and was used for qualitative analyses. The 3-D structure was scanned by searching the Protein Data Bank (PDB)23,24 and ModBase25,26 databases. PDB is the worldwide repository where experimentally determined structures of proteins, nucleic acids, and complex biomolecular assemblies are deposited. These structures are curated and annotated following the standards set by PDB. On the other hand, ModBase is a database of protein structures that have been developed by computational approaches and validated by statistically significant sequence alignment and model assessment.
Qualitative characterization of the ∑ list
Different structural and molecular criteria that facilitate prioritizing therapeutic targets in pathogens were assessed for the ∑ list. 27 This involved the prediction of subcellular location, functional analysis, protein network analysis, broad spectrum analysis, and druggability analysis of the ∑ list.
Subcellular localization analysis
Subcellular localization analysis of a protein reveals whether that protein is suitable as a drug or a vaccine target. Cytoplasmic proteins can work better as drug targets while surface membrane proteins can be used as targets for vaccine development. 28 PSORTb v3.0 server was used to predict the subcellular localization of the proteins, 29 and the results were further crosschecked with predictions obtained from CELLO v.2.5,30,31 TOPCONS, 32 and TMHMM. 33 PSORTb uses 11692 proteins of known localization from bacteria and archaea as a training set to sort out the location of a given protein. CELLO contains 9033 protein sequences as benchmark datasets for the prediction of protein localization using a support vector machine (SVM) method. TMHMM is based on hidden Markov model, and experimental evidence shows that it correctly predicts 97–98% of the transmembrane helices.
Functional analysis
The functions of the hypothetical proteins in the ∑ list were predicted using InterPro online tool. 34 InterPro classifies hypothetical proteins into families and predicts domains and important sites by integrating various protein signature recognition methods and databases.
Broad spectrum analysis
A BLASTp search against a wide range of pathogenic bacteria with an expected threshold value of 0.005 was used to analyze proteins in the ∑ list for the identification of broad spectrum targets. A total of240 disease-causing bacteria from different genus were used in the broad spectrum analysis. 35 From the homology analysis against each of the pathogen, it is speculated that close homologs present in maximum number of pathogens are better likely to be promising broad spectrum targets.
Interactome analysis
A network of protein-protein interaction was constructed for each of the ∑ listed proteins using STRING 9.1.36,37 STRING constructs protein-protein interaction networks based on experimental data, gene-based analysis (neighborhood, gene fusion, co-occurrence, and co-expression), curated pathways database, and various protein interactions databases. High confidence interactors with score greater than or equal to 0.700 alone were included in the protein network. To minimize false positives and false negatives, all interactors with low as well as medium confidence scores were removed from the network.
Druggability analysis
To be druggable, a target must have the potency to interact with drug or drug-like molecules with high affinity. In this study, the draggability potential of each protein of the ∑ list was assessed by DrugBank. 38 Currently, DrugBank is a huge and comprehensive collection of drugs with the target information. This database comprises 6816 experimental and FDA-approved drugs, 4326 drug targets, and 169 drug enzymes/carriers. In DrugBank, each of the ∑ list targets was explored for similar therapeutic targets with the same biological function. Degree of homology was evaluated using the BLASTp program with an expected value of 10-05. Presence of targets from the ∑ list in DrugBank with the same biological function acts as evidence for their druggable property. 39 In contrast, their absence suggests the novelty of the targets, and therefore they are classified as “novel targets.” 40
Ranking the druggable therapeutic targets based on quantitative characteristics
The effectiveness of a putative target may depend on its degree of essentiality for the survival of the pathogenic organism under diverse environmental conditions. Some targets may prove essential for a limited number of physiological conditions, whereas others may prove essential irrespective of environmental conditions. The number of homologs found in DEG and the number of interactors of the particular target are the two cardinal factors that determine the degree of essentiality. Moreover, the number of available drugs present in DrugBank also determines the druggability of a particular therapeutic target. Therefore, we ranked the therapeutic targets by calculating the product of the number of homologs found in DEG, the number of interactors of the target protein, and the number of drugs available in DrugBank. The drug target score was calculated by dividing the product by 100, and the putative therapeutic targets were ranked according to the scores.
Results
In the present study, we identified potential therapeutic targets in M. hominis employing comparative and subtractive genomic analyses of metabolic pathways. We used a systematic hierarchical approach that involved various computational tools utilization, databases search, and drug target prioritization analysis (Fig. 1).
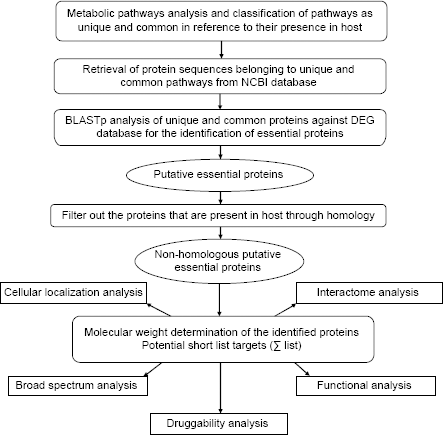
Schematic representation of workflow for the identification of therapeutic targets.
Comparison between M. hominis and H. sapiens metabolic pathways identified five unique pathways and 35 common pathways
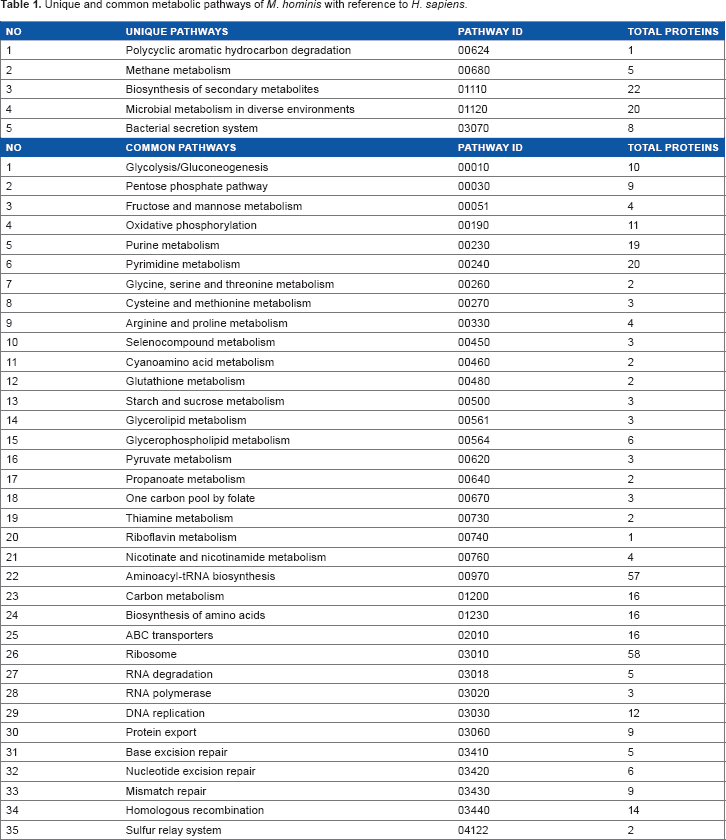
Primary information about the metabolic pathways of M. hominis and humans was retrieved from the KEGG database. Currently, the KEGG database contains information about 40 metabolic pathways for M. hominis (Table 1). Comparison with human pathways revealed five pathways containing 36 proteins as unique to M. hominis while the remaining 35 pathways containing 197 proteins as common to M. hominis and humans. Thirty-five out of 36 proteins of unique pathways are also present in common pathways. After removing redundant proteins, a total of 198 protein sequences were obtained from the NCBI database.
Unique and common metabolic pathways of M. hominis with reference to H. sapines.
Homology search revealed 179 putative essential proteins for M. hominis.
Proteins that are required by pathogenic microorganisms to survive are called essential proteins. Essential proteins are prime targets for drugs and vaccine development. To identify the essential proteins, 198 proteins of common and unique pathways were compared against the DEG database. Out of 198 input proteins, 179 proteins were found to be essential for the pathogen. The distribution of the 179 identified essential proteins in each of the 22 bacteria of DEG is presented in Figure 2.
Comparison of M. hominis genome against DEG. The height of the bars indicates the number of hits on other genomes.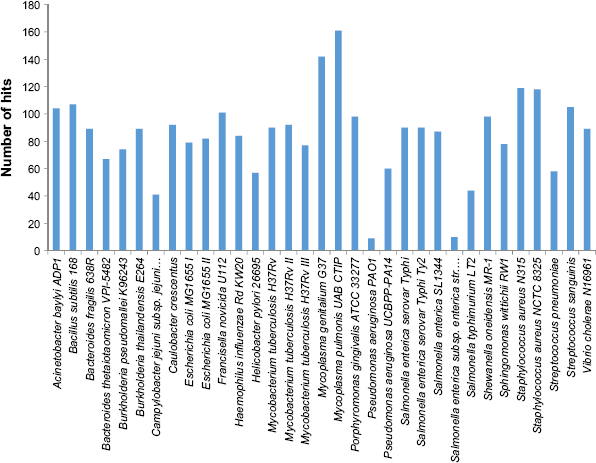
Comparative analysis of the essential proteins revealed 59 of them as non-host
The aim of the non-homology analysis was to identify pathogen-specific proteins that are nonhomologous to the host. This step is important to avoid undesirable cross-reactivity of the drug arising from its binding to the active sites of the homologous proteins in the host. Out of 179 proteins, BLAST search of essential proteins against non-redundant database of H. sapines identified only 59 proteins as non-host but essential proteins.
Exclusion of high MW proteins generated a list of 57 putative therapeutic targets
Proteins of low MW are preferable as drug targets because of their solubility and ease of purification. By MW analysis through online tools and literature study, 57 out of 59 proteins having MW below 110 kDa were short listed (∑ list) for the qualitative characterization. Although no experimentally solved 3-D structure was found for the ∑ listed proteins, computationally annotated 3-D models were available for 16 proteins of the ∑ list in the ModBase database.
Cellular localization analysis mapped the proteins of ∑ list to different cellular locations
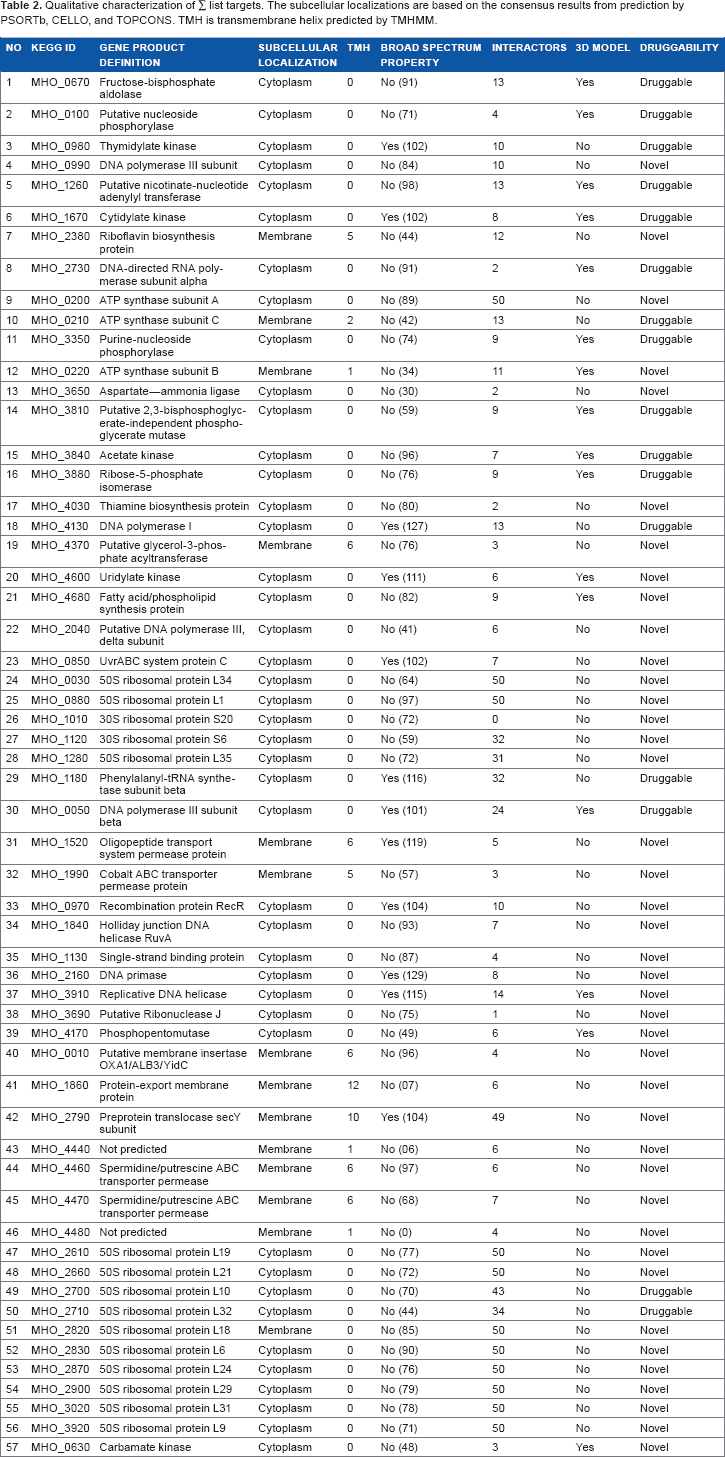
Target proteins found in the cytoplasm can be used as potential drug targets, whereas extracellular and membrane-bound proteins can work as probable vaccine targets. 28 Various online tools were used for the prediction of subcellular localization and membrane topology. Based on the localization score, PSORTb predicted the location of 32 targets in the cytoplasm and 13 in the membrane. However, 12 proteins could not be mapped to any cellular location with this software. CELLO predicted 43 targets in the cytoplasm and 14 in the membrane (Table 2). This tool was able to predict the location of 12 targets previously uncharacterized by PSORTb, and the result of CELLO was in agreement with TOPCONS prediction as well. PSORTb prediction varied with CELLO and TOPCONS in the case of only one protein, MHO_3910. TMHMM predicted the number of transmembrane helixes (TMH) in membrane proteins.
Qualitative characterization of ∑ list targets. The subcellular localizations are based on the consensus results from prediction by PSORTb, CELLO, and TOPCONS. TMH is transmembrane helix predicted by TMHMM.
Biological functions were assigned to seven hypothetical proteins
Nine proteins of the ∑ list with KEGG ID MHO_0100, MHO_1260, MHO_3810, MHO_4370, MHO_2040, MHO_3690, MHO_0010, MHO_4440, and MHO_4480 were hypothetical. Seven of them could be annotated using the InterPro online server (Table 2). MHO_0100 was predicted to have a nucleoside phosphorylase domain with a catalytic role in the nucleoside metabolic process. MHO_1260 is a probable member of the nicotinate-nucleotide adenylyl-transferase protein family having a role in the NAD biosynthetic pathway. MHO_3810 probably plays a role as phosphoglycerate mutase 2,3-bisphosphoglycerate-independent enzyme in glucose metabolism. MHO_4370 and MHO_2040 were recognized as glycerol-3-phosphate acyltransferase (PlsY) and DNA polymerase III (delta subunit) type proteins, respectively. MHO_3690 protein was predicted to be beta-lactamase-like protein having RNA- and metal ion-binding potential. The MHO_0010 was recognized as membrane insertase YidC/Oxa1 (C-terminal) having a pivotal role in protein insertion into the membrane. InterPro could not predict any function for MHO_4440 and MHO_4480 as no homolog was found in the database.
Comparison of proteomes of 240 pathogens identified 12 broad spectrum targets
BLASTp homology search for proteins in the ∑ list against the whole proteome of each of the 240 bacterial pathogens identified ideal broad spectrum targets. This comparative sequence analysis revealed 12 proteins having homologs in more than 100 pathogens, whereas homologs were found in more than 50 pathogens for 33 proteins present in the ∑ list. ∑ listed proteins exhibit maximum homology to the proteome of Mycoplasma pathogens like M. capricolum, M. gallisepticum, M. genitallium, M. penetrans, and M. pneumoniae. Proteins having homologs in more than 100 pathogens were considered as broad spectrum target candidates (Table 2).
Interactome analysis identified 23 targets having maximum interactors
Protein-protein interactions are important determinants of protein function. To evaluate the functional importance of ∑ listed targets in the metabolic network, analysis on protein interaction network was performed using the STRING database. Target protein interacting with more proteins is considered as metabolically important active protein, which can act as an appropriate drug target.41,42 Out of 57 input proteins, 12 were found to have less than five interactors and 23 to have more than 10 interactors (Table 2).
DrugBank database search identified 16 druggable targets
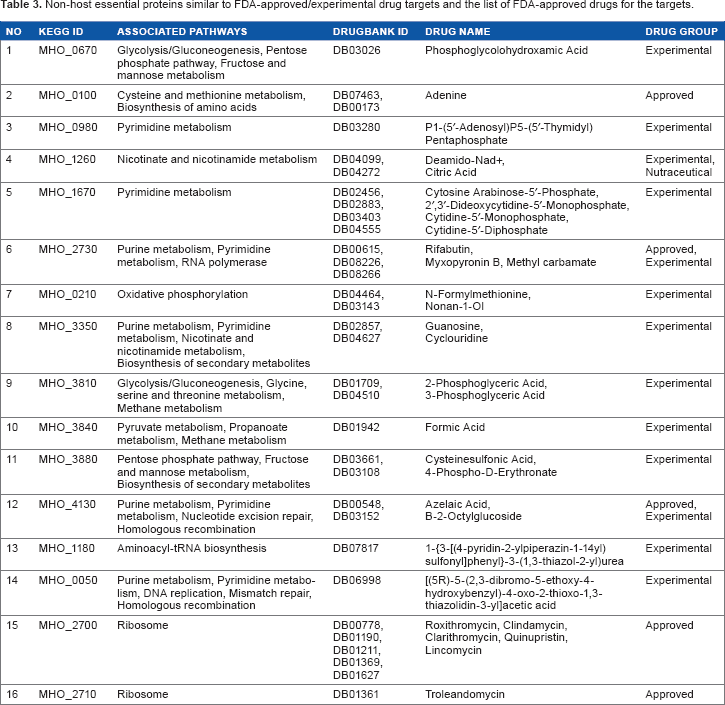
The probability of being druggable of the potential targets can be evaluated by sequence similarity search against the targets from DrugBank.38,39 BLASTp search against DrugBank targets with FDA-approved drugs, nutraceuticals, and experimental drugs revealed that 16 targets in the ∑ list are homologous to DrugBank targets (Table 3). Five targets were found to have homologies to FDA-approved drug targets. Among them, MHO_2700 is homologous to a known target (50S ribosomal protein L10) and has five approved drugs against it, named roxithromycin (DB00778), clindamycin (DB01190), clarithromycin (DB01190), quinupristin (DB01211), and lincomycin (DB01369), which are used to treat Shigella infection. Other four FDA-approved drug targets have one approved drug against each. Proteins in the ∑ list that did not hit with DrugBank database are novel therapeutic targets for which new drug and vaccines can be developed.
Non-host essential proteins similar to FDA-approved/experimental drug targets and the list of FDA-approved drugs for the targets.
Ranking of the drug targets suggests that 50S ribosomal protein L10 has the highest potential to be an effective drug target
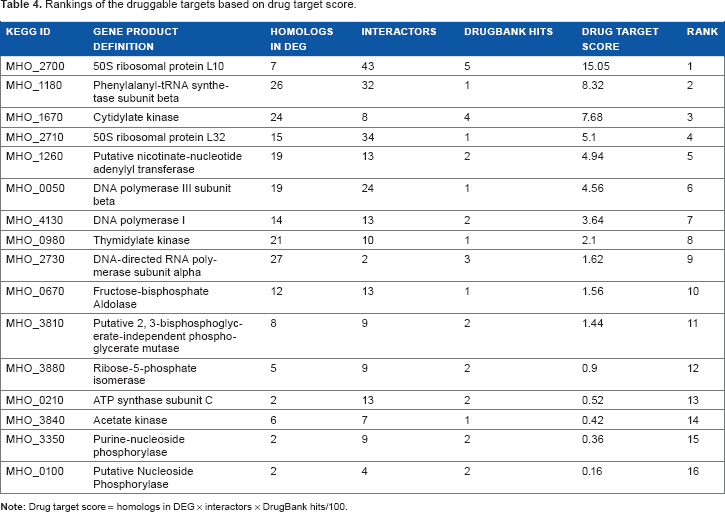
It is useful to rank the putative therapeutic targets based on their quantitative values in order to decide which target has a higher probability of being effective in laboratory experiments. We ranked the putative therapeutic targets based on the number of homologs found in DEG, the number of interactors of the target protein, and the number of drugs available. The ranking of the putative drug targets is presented in Table 4 and showed that 50S ribosomal protein L10 has the highest potential to be an effective therapeutic target. Cytidylate kinase, 50S ribosomal protein L32, and putative nicotinate-nucleotide adenylyl transferase also fall in the group of top five putative therapeutic targets.
Rankings of the druggable targets based on drug target score.
Discussion
The increasing trend of bacterial resistance to antibiotics is posing an imminent public health concern. Researchers around the world are giving attention to find new drug targets so that bacterial infections can be prevented. A vast array of omics data and the availability of various computational tools have speeded up the therapeutic target identification process. The potential of a protein to be a therapeutic target depends on two factors, essentiality and absence in the host. Essential proteins are required for bacterial survival and blocking them inhibits bacterial growth. Non-homologous proteins are preferred because they reduce the probability of side effects. In this study, we extracted metabolic data from KEGG database and used different bioinformatics and computational databases and tools for the identification of appropriate therapeutic targets of M. hominis. Systematic computational analyses revealed 57 possible drug targets in M. hominis; among them 16 are druggable targets, which have homologs in DrugBank. We further utilized different drug prioritization parameters to make a short list of potential drug and vaccine targets. Predicted drug targets belong to a diverse range of cellular activity and fall mostly into ribosome synthesis, pyrimidine metabolism, homologous recombination, DNA replication, and ABC transporter pathways (Fig. 3).
Distribution of putative therapeutic targets in their associated pathways. The percentage distribution of other pathways ranges from 0 to 4.5.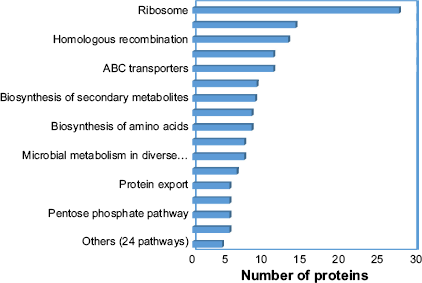
The assembly of bacterial ribosomes has been considered prominently as a potential target for antibacterial drugs. 43 We identified 16 potential drug targets from the ribosome synthesis and assembly pathway. Among them, two are druggable targets that include 50S ribosomal protein L10 and L32. 50S ribosomal protein L10 interacting with acidic L7/L12 proteins constitutes the ribosomal stalk. This stalk is involved in the binding of elongation factors EF-Tu and EF-G and plays a crucial role in activating the GTPase center. This target is highly similar to an already available drug target with five FDA-approved drugs used to control SHIGELLA infection. Our study revealed that 50S ribosomal protein L10 holds the highest promise to be an effective drug target (Table 4). Hence, the potential of it as a drug target remains open for experimental validation. 50S ribosomal protein L32 is a structural constituent of ribosomes. This target was found to have a homologous target in DrugBank for an FDA-approved drug called troleandomycin. However, M. hominis is intrinsically resistant to this antibiotic because of mutations in 23S rRNA, which has stimulated the search for other drug targets. 10
De novo nucleotide synthesis is crucial for the successful growth of bacteria in human blood. 44 Nucleotides synthesized in purine and pyrimidine metabolic pathways are important substrates not only for DNA synthesis but also for DNA repair. We have found five and eight drug targets for purine and pyrimidine metabolism, respectively. DNA polymerase I (Pol I) and DNA-directed RNA polymerase subunit alpha (RpoA), which is involved in nucleotide metabolism as well as other important metabolic pathways, have homologs in DrugBank. Azelaic acid, an approved FDA drug, can be used to treat M. hominis infection by inhibiting the activity of Pol I. This drug is currently being used as an inhibitor of Pol I in ESCHERICHIA COLI. RpoA activity can be blocked by an approved drug, rifabutin, which is highly specific to bacterial RNA polymerase.
In the last few years, aminoacyl-tRNA synthetases (AaRS) have drawn much attention as therapeutic targets because of their crucial roles in protein synthesis and conservation across different pathogens. 45 Here, we highlighted phenylalanyl-tRNA synthetase subunit beta (PheT) as a therapeutic target, which showed considerable homology with a target in DrugBank database. Phenyl-thiazolylurea-sulfonamide, a successful inhibitor of phenylalanyl-tRNA synthetase (Phe-RS), can be tested in the laboratory to determine its efficacy against M. hominis. 46
Glycolysis/gluconeogenesis is perceived as a promising target for new drugs against bacterial pathogens because many of the proteins involved in this pathway are significantly different from human proteins. Here, we identified two proteins of the glycolytic pathway as potential therapeutic targets—one is fructose-bisphosphate aldolase (Fba) and the other is a hypothetical protein, MHO_3810, which is found to have 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (iPGM) activity. Fba catalyses an important reversible reaction required for both glycolysis and gluconeogenesis. 47 Previously, this enzyme has been reported as a target of antifungal and antiprotozoal drugs. Downregulation of iPGM using RNAi resulted in embryonic and larval lethality in Caenorhabditis elegans. 48
In addition to druggable targets, we have also identified several other targets that are involved in crucial bacterial metabolic pathways. Bacterial protein secretion system pathway modulates biotic association as well as pathogenicity. Therefore, proteins from the bacterial secretion system were identified as drug targets in many bacteria.49,50 This secretion system of M. hominis includes eight proteins. Among them three have been proposed as potential therapeutic targets in our study. The proposed three targets are SecD, SecY, and YidC/Oxa1 family membrane proteins, and this result is consistent with two previous studies.51,52 Extensive research is going on in the development of protein secretion inhibitors like salicylidene acylhydrazides 53 and 2-imino-5-arylidene thiazolidinone. 54 Earlier studies reported that ABC transporters play an important role in bacterial physiological processes such as the import of important nutrients required for bacterial growth 55 and export of toxic substances outside of the cell. 56 Here, we have reported six ABC transporters as novel therapeutic targets. These transporters can be used for the development of antibacterial vaccines. 57
Conclusion
This study has identified several proteins that can be targeted for effective drug development. Since some of the identified drug targets play important roles in metabolism, a synchronized approach to develop new drugs would be promising to control M. hominis infection. In addition, results of this study can bring a substantial advancement to test the effectiveness of the currently available drugs. Further wet lab experiments are essential to validate the obtained findings.
Author Contributions
Conceived and designed the experiments: MSH and MMP. Analyzed the data: MMP and MR. Wrote the first draft of the manuscript: MMP. Contributed to the writing of the manuscript: MR. Agree with manuscript results and conclusions: MMP, MR, and MSH. Jointly developed the structure and arguments for the paper: MMP and MR. Made critical revisions and approved final version: MSH. All authors reviewed and approved of the final manuscript.