
Abstract
Fabry disease is an X-linked lysosomal storage disorder that is caused by a deficiency in the enzyme α-galactosidase A. It results in a progressive multi systemic disorder with major organ involvement (principally renal, cardiac and cerebrovascular) as well as peripheral and autonomic nervous system leading to a poor quality of life, and early death. Enzyme replacement therapy with α-galactosidase A has been used to treat Fabry disease since 2001. Two preparations of the enzyme are available: agalsidase alfa, produced in a human cell line, and agalsidase beta, produced in Chinese hamster ovary cells. Both products have a similar composition, mechanism of action and pharmacokinetic profile however major differences exist in the recommended dose, immunogenicity and rate of adverse reactions. Several clinical trials with the two enzyme preparations have assessed clinical efficacy with respect to impact of treatment on kidney function, cardiomyopathy, pain control and quality of life. Both preparations appear to be broadly equivalent. It has not yet been established that early initiation of enzyme replacement therapy (ERT) can prevent the emergence of disease manifestations. This review will cover the main aspects of clinical safety and efficacy of ERT for Fabry disease.
Keywords
Introduction
Fabry disease (OMIM 301500) is an X-linked hereditary disorder caused by a deficiency in the lysosomal enzyme, α-galactosidase A (α-galA). 1 As a consequence, the enzyme substrate, globotriaosylceramide (Gb3), accumulates in various cell types and organs throughout life leading to multi-organ pathology that most severely affects the kidneys, heart, peripheral and autonomic nervous system, and cerebrovascular system, and results in premature mortality. The incidence of Fabry disease has been estimated to be about 1 in 117,000, 2 but newborn screening programs have revealed a higher incidence (from 1: 3,100 in Italy to 1: 1,250 in Taiwan male births), 3 with a high proportion of GLA gene mutations that would likely result in a later onset and less severe phenotype. 3 There are several recent reviews.4–7
The initial signs and symptoms of Fabry disease in males typically appear during childhood and adolescence. They include neuropathic pain crises (ie, acroparesthesia), exercise and heat/cold intolerance, angiokeratomas, disordered sweating, tinnitus and gastrointestinal problems, including diarrhea, constipation, abdominal pain, and vomiting.8,9 Patients are frequently misdiagnosed, and the correct diagnosis may be delayed. 10 Many patients present whorl-shaped corneal opacities that usually do not affect vision. 11 These changes are highly specific for Fabry disease, although similar abnormalities may occur in subjects taking drugs including amiodarone. Major organ involvement typically occurs in adult patients and includes kidney dysfunction progressing to end-stage renal disease (ESRD), 12 cardiomyopathy, cardiac arrhythmias and valve dysfunction, and a high incidence of stroke and transient ischemic attacks. 13 These major organ pathologies account for most of the premature mortality in Fabry disease.14,15 Chronic kidney disease occurs in nearly all men with Fabry disease.12,16,17 Cardiac disease, including progressive development of left ventricular hypertrophy (LVH), valve thickening, and conduction abnormalities, is common. 18 Cerebrovascular complications may include early stroke, transient ischemic attacks and cerebral vasculopathy with cerebral hemorrhages.19,20 The most common symptoms in children are neuropathic pain (63% of boys, 46% of girls), hypohidrosis (34% of boys, 14% of girls), and gastrointestinal problems (33% of boys, 19% of girls). 9 Major organ involvement has also been reported in children and adolescents, including reduction in GFR and proteinuria,21,22 LVH, 23 and stroke. 24 Quality of life for affected adults and children is reduced and patients are at risk of depression.25,26
Despite an X-linked inheritance, female heterozygotes present a highly variable spectrum of the disease ranging from almost asymptomatic to severely affected.27,28 Signs and symptoms of Fabry disease are reported in females during childhood and adolescence, 9 but compared with male patients they typically emerge later in life and with less severity. End stage renal disease, severe cardiomyopathy, and stroke are all seen in women.29,30 Skewed X-chromosome inactivation is thought to be responsible for the expression of Fabry disease in women but may not be sufficient to explain the variable presentation.31.
Management of Fabry Disease
The management of Fabry disease was purely supportive and focused on the symptomatic treatment. However, the development of biotechnology has allowed the production of human α-galactosidase A for ERT of Fabry disease and genuine opportunities to alter the natural history of the disease have emerged. Fabry disease is a multi system disorder affecting adults, children and families. The importance of a multi-disciplinary approach to management has been emphasized4,7 and patients should be assessed by pediatricians or adult physicians who are able to assess the impact of the disease across the full range of organ systems including its psychosocial aspects. Elsewhere we have proposed ‘Goals of Management’ which can be applied in a clinical setting. 7 The Mainz Severity Score Index (MSSI) is an instrument for quantifying the overall severity of the signs and symptoms of Fabry disease. 32 The MSSI assigns scores based on the presence and severity of signs and symptoms in 4 areas: general, neurological, cardiovascular, and renal. The multi-disciplinary team also has a key role in optimizing supportive therapies. This includes management of pain, hypertension, renal failure, proteinuria, cardiac disease and cerebrovascular disease.
The studies of ERT in Fabry disease have assessed its impact on both the biochemical and pathological parameters as well as on clinical measures. 6 Biochemical studies are hampered by the lack of reliable and clinically relevant biomarkers of the disease. From the pathophysiologic point of view, storage of Gb3 represents the initial metabolic event that may start even before birth. 33 Although the storage of Gb3 has been considered as the primary pathologic event, no direct correlation has been demonstrated between Gb3 storage and disease progression. Thus, Gb3 does not represent a good biomarker for Fabry disease progression or the evaluation of response to treatment.34,35 Recently, Aerts and collaborators reported that globotriaosylsphingosine (lyso-Gb3), a Gb3 derivative, is consistently increased in plasma of male Fabry patients and, although to a lesser extent, in symptomatic females. 36 Plasma lyso-Gb3 is an independent risk factor for development of cerebral white matter lesions in male patients and left ventricular hypertrophy in females. Disease severity correlates with exposure to plasma lyso-Gb3. 37
Studies of the impact of ERT on clinical aspects of the disease have examined effects on pain, kidney disease, cardiac parameters and quality of life. 6 The blood brain barrier prevents ERT from accessing the central nervous system. Additional studies on other disease manifestations (eg, hearing loss, sensory disturbances and GI manifestations) have also been reported. The rarity of the condition means that the studies are frequently small and lacking in rigorous design. The measured outcomes are difficult to measure objectively, reliably and reproducibly; furthermore, these outcomes are at best surrogate markers of the severity of the disease. The paucity of detailed natural history data makes retrospective studies (which comprise the bulk of the literature) more difficult to interpret.
The Enzyme Products: Agalsidase Alfa (Replagal™) and Agalsidase Beta (Fabrazyme™)
Two different products are available for the treatment of Fabry patients: agalsidase alfa 0.2 mg/kg every other week (EOW) (Replagal®, Shire Human Genetic Therapies, Inc. (Shire HGT), Cambridge, MA, USA), which is produced by gene activation in a human fibroblast cell line, and agalsidase beta 1.0 mg/kg EOW (Fabrazyme®, Genzyme Corporation, Cambridge, MA, USA) which is produced by recombinant techniques in Chinese hamster ovary cells. Both products were initially approved in 2001 and are now approved in many countries around the world (Europe, Canada, Australia, Latin America, and Japan). As of September 2010, only agalsidase beta has been approved in the U.S.A.
The amino acid sequence of both drugs is identical to the endogenous human enzyme. However the glycosylation pattern may differ as the carbohydrate structure is added by systems that may differ between human and hamster. Biochemical and pharmacokinetic properties are similar, including cellular uptake and functional correction in vitro in cultured fibroblasts from Fabry patients. 38 A detailed description of these aspects is given elsewhere.5,39
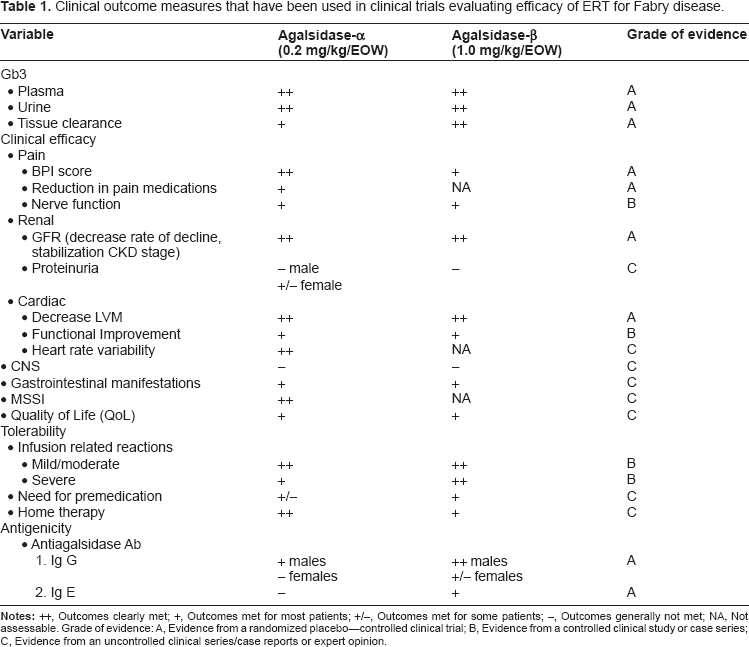
Table 1 lists the outcome measures used in various trials. The two products are assessed with regard to the extent to which these outcomes are achieved in clinical trials.
Clinical outcome measures that have been used in clinical trials evaluating efficacy of ERT for Fabry disease.
Clinical Efficacy of Agalsidase Alfa: Randomized Clinical Trials
Randomized, placebo-controlled clinical trials of each preparation have been conducted.
Agalsidase alfa was evaluated in a randomized double-blind, placebo-controlled clinical trial of 26 adult male patients. 40 The entry criteria were confirmed Fabry disease and the presence of neuropathic pain. Patients were randomized to placebo or agalsidase alfa 0.2 mg/kg EOW for a period of 6 months, and the severity of neuropathic pain was assessed with the Brief Pain Inventory (BPI). BPI “pain at its worst” score was significantly reduced by 6 months of agalsidase alfa relative to placebo. The patients in the placebo group experienced a similar reduction in BPI “pain at its worst” score 6 months after starting agalsidase alfa in the open-label extension study. 41
Creatinine clearance declined in the placebo group from 100.6 ± 12.2 mL/min/1.73 m2 at baseline to 84.5 ± 10.6 mL/min/1.73 m2 after 6 months but remained stable in the agalsidase alfa group. (P = 0.02 for comparison between groups). This study was continued as an open-label extension study for an additional 4 years 42 and the eGFR of patients with normal (N = 12) or only mild renal impairment (CKD stage 1–2, N = 8) at baseline did not change significantly. The four patients with more severe kidney disease (CKD satge 3, baseline eGFR between 30 and 60) showed a further decrease in mean eGFR during the study period. No changes in proteinuria were observed during the study.
In a separate study of fifteen males patients with Fabry disease and left ventricular hypertrophy, 6 months of agalsidase alfa reduced left ventricular mass (LVM) in adult men compared with placebo. 43 A summary analysis of three trials of the effects of agalsidase alfa on kidney function as assessed by measured GFR 44 demonstrated that GFR was stabilized in patients with mild-to-moderate kidney dysfunction at baseline and that this benefit was primarily seen in patients with baseline proteinuria <1 g/day.
Clinical Efficacy of Agalsidase Beta: Randomized Clinical Trials
Two randomized clinical trials have assessed the efficacy of agalsidase beta.
In a group of 56 men and 2 women with Fabry disease, Eng and colleagues reported significant reductions in pain scores during a 6-month, double-blind study, but baseline scores were low and similar significant reductions were also seen in the placebo group. 45
Therapy with agalsidase beta was shown to clear Gb3 deposits from skin and heart after 6 months, 45 and appeared to stabilize kidney function during the 4-year, open-label extension study. 46 In this extension phase Germain and coworkers reported that eGFR declined by about 1.1 mL/min/1.73 m2/year in 58 adult patients with normal pre-treatment eGFR treated with agalsidase beta for a mean of 52.2 months. 46 Baseline proteinuria >1 g/day was significantly associated with continued progressive loss of eGFR during the study. Median proteinuria did not change during the study.
The same randomized study has also shown that treatment with agalsidase beta was associated with delayed time to first major clinical event a composite of renal, cardiac, or cerebrovascular event or death, hazard ratio (HR = 0.47, P = 0.06). 47 Although the majority of events in both the placebo and agalsidase beta groups were renal (ie, a 33% increase in serum creatinine), no change in mean eGFR, serum creatinine, or proteinuria was seen in either group during the median 18.5-month study period. This benefit was most prominent in patients with baseline proteinuria <1 g/day.
Agalsidase Alfa: Other Clinical Studies
Kampmann and colleagues reported a retrospective, blinded assessment of echocardiograms obtained from male and female patients with Fabry disease who had been enrolled in previous clinical trials. 48 Fourteen subjects (9 men and 5 women) had LVH at baseline and 31 patients did not have baseline LVH. Treatment with agalsidase alfa for 12 or 36 months was associated with reduced LVM in patients with Fabry disease with baseline LVH, and it appeared to stabilize LVM in those patients without baseline LVH. In women, BPI “pain at its worst” scores were significantly reduced after 1 year of treatment, and this benefit was sustained through a 4-year study. 49 Evaluation of the effect of agalsidase alfa on peripheral nerve function has yielded inconsistent results. Schiffmann and colleagues reported an improvement in hot and cold sensation in the foot, as well as sweating during up to 3 years of agalsidase alfa therapy but no increase in intraepidermal innervation density, suggesting that epidermal nerve fibers do not regenerate during agalsidase alfa treatment.50,51
The effect of ERT on cerebral vascular perfusion and function was evaluated in two 6-month randomized controlled trials with agalsidase alfa.52–54 Several studies have demonstrated significant functional changes in the vasculature of the brain, but no effect on the incidence of stroke has been demonstrated and stroke has been observed in some patients during treatment with agalsidase alfa or beta in clinical trials.42,55 Increases in the number of white matter lesions detected by MRI during agalsidase alfa have been reported.56,58
In a small study of adult male Fabry disease patients treatment with agalsidase alfa led to an improvement in hearing. 59 Palla and colleagues studied 37 patients treated with agalsidase alfa and reported an improvement in vestibular function within the first year of ERT while auditory function did not improve during the 5-year study. 60 These small changes are probably insufficient to affect quality of life.
Baehner and colleagues reported the results of a single-center, open-label clinical study of agalsidase alfa in 15 adult women with symptomatic Fabry 61 The study was continued with additional female patients (total n = 36) for a total treatment time of 4 years. 49 Mean baseline eGFR was 91.0 ± 31.2 mL/min/1.73 m2 and did not change during the 4 years of treatment with agalsidase alfa. Proteinuria (>300 mg/24 h) was present in 11 patients at baseline (mean = 858 ± 751 mg/24 h) and in this subgroup, mean proteinuria was significantly reduced to 339 ± 230 mg/24 h after 4 years of treatment (P ≤ 0.01). LVM, as assessed by echocardiography, was elevated at baseline in 25 of 36 patients and was significantly reduced after 1 year of agalsidase alfa in this subgroup as well as in those patients with normal baseline LVM. These reductions were accompanied by improvements in BPI pain scores which persisted through 4 years of treatment.
Gastrointestinal disturbances, primarily abdominal pain and diarrhea, are common in men, women and children with Fabry disease.9,62,63 A single study of 11 patients treated with agalsidase alfa for 6 months observed that the frequency and severity of abdominal pain were significantly reduced and 5 of 6 patients with baseline diarrhea experienced an improvement. 64
In a study including 30 patients (23 males, 7 females) who were treated for 1 to 5 years with agalsidase alfa, total MSSI scores were significantly improved compared to baseline (P < 0.0001). 65 Whybra and colleagues reported that total MSSI scores in a group of 36 women were significantly reduced after 1 year of agalsidase alfa and remained significantly reduced through 4 years of treatment. 49 Improvements in the neurological and cardiovascular subscores were responsible for most of the improvement.
Registry studies are not well controlled and the data is often incomplete. However long term data are now available from large numbers of patients and they suggest that long term agalsidase alfa therapy may be beneficial. 66
Agalsidase Beta: Other Clinical Studies
The role of baseline severity of renal dysfunction in relation to ERT response was studied by Breunig et al 67 in a group of 26 patients. Baseline eGFR categorized as above or below 90 mL/min/1.73 m2 had no apparent effect on the change in eGFR during 2 years of agalsidase beta. However patients with higher levels of baseline renal dysfunction responded less well to ERT.
The effect of agalsidase beta on LVM has been investigated in multiple open-label clinical studies. Two of the studies reported no change in LVM after 1 or 2 years of agalsidase beta treatment.68,69 The remaining studies found a significant decrease in LVM in either the entire study population or in one or more subgroups.70–74 Patients with myocardial fibrosis respond less well.
Improved sensory perception in Fabry patients treated with agalsidase beta, particularly with respect to heat pain thresholds, were reported by Hilz et al. 75 These authors found improvements in vibratory, cold, and heat-pain detection thresholds after 18 or 23 months of agalsidase beta, but some patients exhibited no change or worsening of threshold during treatment, suggesting that they had experienced irreversible changes prior to initiation of ERT.
Paediatric Studies of ERT in Fabry Disease
Two studies of agalsidase alfa in children have been reported. Ries and co-workers treated 19 boys and 5 girls initially for 6 months 24 and extended to 4 years. 41 Agalsidase alfa was generally well tolerated. Sweat function was assessed by quantitative sudomotor axon reflex testing in 13 patients and showed an increase in sweat volume from 0.48 μL/mm2 at baseline to 0.73 μL/mm2 at 6 months (P = .06). All time-domain indices of HRV were improved in boys treated with agalsidase alfa. 24 The effect on HRV persisted throughout the extended period, and significant improvement in BPI “pain at its worst” scores was observed after 1 year of ERT. A similar open-label study was conducted by Ramaswami and colleagues, 76 who treated 9 boys and 4 girls for 23 weeks. BPI pain scores tended to decrease in this clinical trial that was mainly designed to study safety in children.
One study of agalsidase beta in children has been reported. Wraith et al 77 treated 14 boys and 2 girls for 48 weeks and reported a decrease in the prevalence of postprandial pain and vomiting compared with baseline.
Comparative Studies of the 2 Products
Comparison between the two products is difficult because of the different cell source of the enzymes, the different recommended dose based on different objectives in the preclinical trials and, in addition, because of different primary end points in the clinical trials. The initial strategy for evaluating the efficacy of agalsidase alfa during phase III studies consisted primarily in assessing clinical criteria: effect on neuropathic pain, left ventricular size, and preservation of glomerular filtration rate. On the contrary, the primary end point for agalsidase beta was the renal endothelial clearance of Gb3.
Vedder et al conducted a randomized study comparing agalsidase alfa at its approved dose (0.2 mg/kg EOW) with agalsidase beta at 0.2 mg/kg EOW, a dose that is less than its approved dose of 1.0 mg/kg EOW. 78 Thirteen of the 14 patients who were assigned to agalsidase alfa and had a baseline echocardiogram were classified as having LVH at baseline (median LVM = 288 g). LVM declined by a median 23 g after 1 year of agalsidase alfa and by a median 11 g after 2 years of agalsidase alfa (P value not significant for both years compared with baseline). Treatment failure (defined as progression of kidney disease or cardiac disease, or the occurrence of a new cerebrovascular event) was observed in 5 of 18 patients in the agalsidase alfa group and in 3 of 16 patients in the agalsidase beta group (P = 0.54). Further progression of CNS involvement was seen in 5 of these 8 treatment-failure patients after switching to agalsidase beta at 1.0 mg/kg EOW. In this study, no clear difference in clinical effect was demonstrated between the two enzyme preparations. Furthermore, the design has been criticised, particularly in respect to pooling of the groups of patients treated with agalsidase alfa or agalsidase beta at 0.2 mg/kg every other week. 79
The licensed dose of the two preparations is different and it has been suggested that this may contribute to differences in treatment effect and immunogenicity. Clark et al 80 conducted a pharmacokinetic study of five agalsidase alfa regimens in 18 males with Fabry disease naïve to the drug. The reduction of Gb3 in the different groups of patients was found to be similar, and based on the effects on plasma Gb3 levels, increased dosages or frequency were not superior to the recommended dose of 0.2 mg/kg EOW.
Schiffmann 81 et al reported that in a subgroup of 11 adult male patients an increased dose of agalsidase alfa (0.2 mg/kg weekly) was associated with a reduction in the rate of decline of measured GFR. Lubanda et al 82 have reported the effects of reducing the dose of agalsidase beta to 0.3 mg/kg EOW in a group of 21 adult male Fabry patients who had previously been treated for at least 6 months at the licensed dose of 1.0 mg/kg EOW. Although some patients appeared to tolerate this dose reduction, 30% reported deterioration and disease progression. An ongoing clinical trial in male children with Fabry disease (FIELD study) is exploring whether 2 alternative dosing regimens of Fabrazyme (1.0 mg/kg every 4 weeks or 0.5 mg/kg every 2 weeks) are effective in treatment-naïve pediatric patients without severe disease manifestations. Such studies seem entirely appropriate, given the similarity of the molecular composition of both agalsidase preparations.
Safety and Tolerability
In clinical trials and post-marketing surveillance, the most common drug-related adverse events are infusion-related reactions, with a reported frequency about 10%. These infusion-related reactions typically involve fever, chills, rigors, flushing, nausea, and/or headache and are managed acutely by slowing or stopping the infusion and administering antihistamines. Patients who have experienced an infusion-related reaction are pretreated with an antihistamine and low-dose oral corticosteroids prior to subsequent infusions. Most patients treated with agalsidase alfa can be weaned from these premedications without further reactions.
Agalsidase alfa and beta may be viewed as foreign proteins by the immune system of male patients, and therefore, it is not surprising that anti-agalsidase antibodies are reported in men and boys with Fabry disease. According to data on file with the European Medicines Agency, 24% of men treated with agalsidase alfa develop IgG antibodies and 89% of patients treated with agalsidase beta develop IgG antibodies.83,84 IgG antibodies have not been reported in women treated with agalsidase alfa, 49 but they do occur in adult women treated with agalsidase beta.46,47 To date, IgE antibodies have only been reported in patients treated with agalsidase beta.46,47 IgG anti-agalsidase alfa antibodies are reported to attenuate some of the biochemical responses to agalsidase alfa. For example, the reduction in urinary excretion of Gb3 is less in men with IgG anti-agalsidase alfa antibodies,42,85 and these antibodies may influence the Gb3 storage in skin capillaries although no relation between antibody formation and plasma Gb3 levels or clinical outcomes has yet been established. 86 The effects of anti-agalsidase alfa antibodies on pharmacokinetics or biodistribution of the enzyme have not been described. Although no clear clinical consequences of antibody formation have been demonstrated, it is known that these antibodies have high neutralizing capacities. 87
A single head-to-head trial has been conducted and found that more patients treated with the licensed dose of agalsidase beta (1.0 mg/kg EOW) developed IgG antibodies (8 of 10) than patients treated with the licensed dose of agalsidase alfa (0.2 mg/kg EOW; 4 of 10; P = 0.005). This difference did not appear to be entirely due to the larger dose of agalsidase beta, as patients treated with a lower dose (0.2 mg/kg EOW) of agalsidase beta also developed IgG antibodies at a higher rate (6 of 8 patients). 85
The Current Shortage of Agalsidase Beta
Viral contamination of a Genzyme manufacturing plant in Massachusetts, detected in June 2009, caused an unexpected shortage of imiglucerase (Cerezyme), for Gaucher's disease, and agalsidase beta (Fabrazyme), for Fabry's disease. The virus, vesivirus 2117, interfered with the growth of Chinese-hamster-ovary cells, which are used to produce biologic drugs, leading to a decline in productivity. The virus has not been shown to cause infection in humans. Genzyme has managed the shortage of these very expensive drugs by using existing inventories and developing dose-conservation measures. In response to the shortage of Fabrazyme, FDA has been in discussion with Shire regarding possible options that would allow Fabry patients in the U.S.A. access to Replagal. At this time, individual Fabry patients may access treatment with Replagal under emergency or single-patient INDs based on clinical need as assessed by their treating physician. In Europe, the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) recommends that, in situations where Replagal treatment is available, no new patients should be started on Fabrazyme. 88 The EMA also recommends that for patients receiving a dose of Fabrazyme less than 1 mg/kg every other week, physicians should consider switching to Replagal at licensed dose. 88
Patient Preference
There are many determinants of patient preference and different individuals will give more weight to some aspects than others.
Agalsidase alfa has a shorter infusion time, is generally better tolerated and pre-medication is not usually required. Home infusion is widely practised and is well established for agalsidase alfa89–91 in some countries, notably the UK and Holland.
Following the restrictions to the supply of agalsidase beta dating from June 2009, some patients have accepted a smaller dose of agalsidase beta than they had previously received while others have decided to switch to agalsidase alfa. Patients’ decisions will have been influenced by advice from their physicians, from patient organizations and from recommendations published by regulatory agencies. 88 Patients have been fortunate in having a choice of licensed preparations for an orphan disease.
Patients should be informed of the risk of clinical deterioration if they suspend ERT. They should also be aware that there may be a greater risk of unstable disease if they reduce the treatment dose.
Place in Therapy and Future Prospects
Fabry disease has now become a treatable, rare metabolic disease and, on the basis of the presented data from different studies, the natural progression of the disease may be stabilized if ERT is instituted at an appropriate time. Treatment challenges remain, however:
ERT is not completely effective at reducing disease progression and established organ damage is frequently irreversible. ERT should be instituted before the onset of established organ damage.
ERT does not reach CNS and will not treat or prevent CNS disease.
ERT has not been shown to reduce the risk of cerebrovascular events.
ERT has to be administered intravenously and this is less convenient than oral administration and makes the treatment unavailable for patients who do not have venous access.
Patients receiving ERT should be closely monitored and ancillary care, particularly regarding management of pain, hypertension, and renal, cardiac and cerebrovascular disease, should be optimized. The importance of ancillary care was emphasized by Tahir et al who reported a sustained reduction of proteinuria in patients receiving ERT with agalsidase beta in association with antiproteinuric therapy comprising angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers. 92
The high cost of ERT, not only for Fabry disease treatment but also for other lysosomal storage disorders (Gaucher, Pompe, Hurler, Hunter, Maroteaux-Lamy, and in the near future, Morquio A and Sanfilippo diseases) represents an extremely high burden for health care systems. This cost is an important cause of reduced access to ERT in many countries throughout the world.
Small molecule therapies are also being developed for the treatment of Fabry disease.
These treatments are oral and have the potential to cross the blood brain barrier. Chaperones are small molecules (iminosugars) that are active-site inhibitors which at low doses can promote the normal folding and trafficking of misfolded wild type and mutated proteins. 93 In patients with certain missense mutations or in-frame deletions, protein misfolding may occur, but some residual enzyme activity remains. Molecular chaperones bind to and stabilize the protein and prevent misfolding during the transit from the Golgi to the lysosome. Migalastat hydrochloride was tested in a Phase 2 clinical trial in 18 men and 9 women with Fabry disease. 94 On average, α-galA levels in leukocytes of treated males increased more than 4-fold, and decreases in urine Gb3 levels were seen in the patients with the largest increases in peripheral enzyme activity. Substrate reduction therapy has been proposed as a way to restore the balance between production and degradation of substrate in lysosomal storage diseases. A glucosylceramide synthase inhibitor, N-butylde-oxynojirimycin, has recently been shown to reduce vascular Gb3 accumulation in Fabry knockout mice. 95
Conclusions
The two ERT preparations agalsidase alfa and agalsidase beta appear to be of comparable efficacy in the management of Fabry disease. Both enzymes when used at the approved dose, commenced at an appropriate time in the evolution of the disease, and used with appropriate supportive therapy are capable of stabilizing renal function and also left ventricular hypertrophy and cardiac function. However the impact on long-term survival has not been established. Both products have significant limitations with regard to their efficacy in improving cardiac conduction, proteinuria, and preventing CNS disease and cerebrovascular events. It is likely that advanced disease stage with irreversible organ damage prior to the start of treatment influence outcome. Studies in asymptomatic young individuals may provide information on the effectiveness of ERT in preventing progressive organ disease or disease-related complications.
An important difference is noted with respect to antigenicity and tolerability. Agalsidase alfa is better tolerated and its use is less likely to provoke antibody formation. In addition, the presence of high antibodies may jeopardize the future possibility of switching to a lower (maintenance) dose of ERT without compromising efficacy.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere: G. Pintos-Morell has received honoraria and/or travel expenses from Shire HGT, Genzyme and Biomarin. O. Lidove has received support and/or travel expenses from Shire HGT, Genzyme, and Actelion. A. Mehta has received honoraria, research funding, consultancy fees, and travel expenses from Shire HGT, Genzyme, Actelion, Protalix, and Amicus. The peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.