
Abstract
Fucosidosis is a rare lysosomal storage disease, resulting from a deficiency in an alpha-
Introduction
Fucosidosis is a lysosomal storage disorder caused by mutations in the FUCA1 gene, which encodes alpha-
Case report
An 8-year-old Chinese boy with a 2-year history of abnormal gait and born to non-consanguineous parents, presented to Quanzhou Children Hospital (Quanzhou, China). No family history of neurodegenerative disease was reported, and the patient had an uneventful perinatal history. His psychomotor milestones were normal during the first 8 months of life. However, he developed psychomotor regression after that time. At 1 year and 7 months, he was able to stand without support and walk alone but was prone to falling. In the two years before presenting to the hospital, he had progressive motor deterioration and walked on his toes, which made him fall frequently. He also had language developmental delay and was able to spell only one- or two-syllable words. A pink maculopapular rash was found in the genital area at birth, which extended to the whole body, including palms and soles, with increasing age. This finding was recognized as suspected telangiectasia (Figure 1a–c). The boy had a respiratory infection every 1 or 2 months, even though immunological investigations were normal. Physical examination showed dry skin and slight facial coarseness, with a protruding forehead, arcuated eyebrows, and broad eye gap. His lips were thick and no macroglossia was observed. Muscle tension was high in the lower limbs, especially in the lower extremity, resulting in plantar flexion. Brisk bilateral deep tendon reflexes were found in the lower limbs. His intelligence quotient was <40. Lateral radiology of the cervical, thoracic, and lumbar spine showed oval vertebral bodies and anterior beak-like formations with osteoporosis (Figure 2a–c). The frontal radiology of the pelvis showed irregular femoral capital epiphyses, with possible sclerosis. The acetabular roofs were steep with an angle of approximately 30° bilaterally, and the lower iliac part was constricted (Figure 2d). Echocardiography was normal, and no visceromegaly was found. Head magnetic resonance imaging (MRI) showed T1-hyperintense and T2-hypointense areas in the bilateral pallidum with curvilinear T2-hyperintense areas within lentiform nuclei, a sign sometimes called “eye of the tiger.” Hyperintense regions on T1- and T2-weighted images were observed in symmetric periventricular white matter (Figure 3); however, no atrophy was observed. Screening for inherited metabolic diseases, including Fabry disease, mannosidosis, and mucopolysaccharidoses, was negative. Because of the MRI “eye of the tiger” finding, a mutation in the PANK2 gene was assumed; however, analysis of this gene showed no mutation. Next-generation sequencing indicated a homozygous mutation in the FUCA1 gene, c.393(exon2)T>A, p.Tyr131Stop, which has been described previously. The patient was diagnosed with fucosidosis.

Maculopapular rash on the patient (a–c). Punctiform or speckled papules of a pink or dark-red color on abdomen (a), forearm (b), and palm (c) of the patient. He also had mild morphological features with dry skin, arcuated eyebrows, and broad eye gap.

Radiology of spine (a–c) and pelvis (d). Lateral radiology of the cervical (a), thoracic (b), and lumbar spine (c) demonstrated anterior beak-like formation and oval-shaped vertebral bodies with osteoporosis. Frontal radiology of pelvic showed irregular femoral capital epiphyses and steep acetabular roofs with an angle of approximately 30° bilaterally (d).
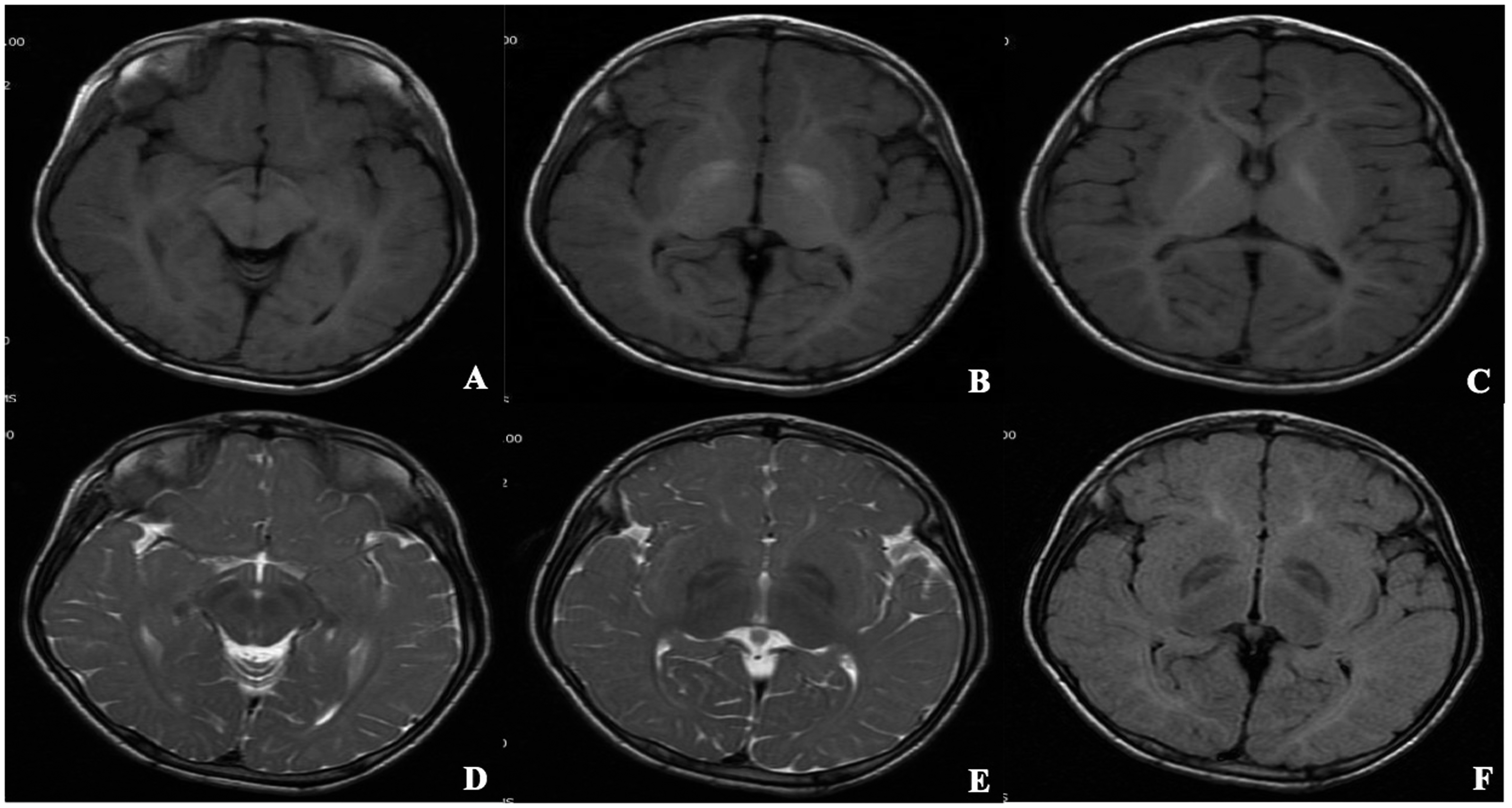
T1-weighted axial (a–c), T2-weighted axial (d, e), and FLAIR (f) MRI. In the bilateral pallidum, axial MRI showed high signal intensity on T1-weighted (a–c) and low signal intensity on T2-weighted and FLAIR images with curvilinear high-signal intensity within lentiform nuclei (d–f). Symmetric hyperintensity in bilateral periventricular white matter on T1- and T2-weighted images was consistent with hypomyelination (b–e). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
All identifying details of the patient’s information have been deleted from the case report, and the identity of the patient cannot be ascertained in any way; therefore, signed consent from the patient or his parents was deemed unnecessary. The patient’s parents consented to publication of the patient’s history.
Discussion
Symptoms of fucosidosis include coarse facial features, visceromegaly, neurologic disorders, dysostosis multiplex, and angiokeratomas. However, it is important to note that symptoms vary considerably. Fucosidosis can be categorized into two types:2,6,7 type 1 begins in early infancy, usually at about 6 months of age, and has a more rapid progression, especially in neurologic deterioration; type 2 initiates before the age of 2 years and progresses more slowly. Individuals with type 1 often die between the age of 5 and 10 years, whereas patients with type 2 might survive to the second decade, although rarely to the age of 30.2,6,7 However, current opinion is that severity of the clinical disorder might exist on a continuum rather than two clear-cut distinct types as considered previously.2,7
No relationship has been found between clinical severity and specific mutation or residual activity of alpha-
The first observed symptom in our patient was telangiectasia. Telangiectasia or angiokeratoma is a skin change observed in fucosidosis. In a review of 77 fucosidosis patients, about 51% presented with angiokeratoma. 7 The presence or absence of angiokeratoma likely depends on the age of the patients examined; angiokeratoma is more likely to be found in older patients. Only about 34% of patients below 10 years of age have angiokeratoma, whereas 88% of patients older than 20 years show the skin change. 7 At the initial patient assessment, angiokeratoma might present as simple telangiectasia and later develop into angiokeratoma. However, some patients have only telangiectasia without angiokeratoma.7,8 In our patient, telangiectasia was found, and it is unknown whether angiokeratoma will appear as time passes. The mechanism underlying the dermatological abnormality is unclear. It is speculated that accumulation of pathologic materials, including a partial breakdown of glycolipids and glycoproteins, leads to apoptosis of endothelial cells, and then these cells continuously regenerate, which results in ecstatic capillaries. 9 An ultrastructure study has found varying degrees of vacuolation in melanocytes, endotheliocytes, and sweat gland cells of skin. 10
The MRI of our patient showed a hypointense globus pallidus with linear hyperintense inside on T2 images. The low signal on T2 in the globus pallidus has been described previously in fucosidosis, 11 although the underlying mechanism is unclear. Several explanations, including cerebral glycolipid and triglyceride deposition and accumulation of calcification or iron following subacute hemorrhage, have been proposed.11,12 The hyperintense view of the T2 images in periventricular white matter in our patient indicated demyelination, which might also affect subcortical white matter, 11 although it was not obvious in our case. One reported case showed increased cerebellar volume in the early stage of fucosidosis, 13 but generalized cerebral atrophy becomes prominent with clinical deterioration. 6 No obvious atrophy was found in our case; however, a follow-up is necessary to monitor changes.
The genetic alteration observed in our patient has been reported previously and is known to result in a stop codon that would truncate the protein at amino acid position 131 and thus affect activity of the alpha-
Conclusion
Fucosidosis is a rare disorder. Telangiectasia or angiokeratoma might be an early indicator but is inconspicuous in the early stage of the disease. Paying attention to the classic symptoms, including visceromegaly, neurological deterioration, dysostosis multiplex, and neuroimaging, is critical for accurate diagnosis. Hematopoietic stem cell transplantation may be a suitable therapeutic approach in the early stage of the disease; thus, early diagnosis is necessary.
Footnotes
Authors' contributions
LW and MY were major contributors in writing the manuscript; JZ and HH analyzed and interpreted the data; and SH and TT helped to collect the patient data. All authors read and approved the final manuscript.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This publication was funded by a grant from the Natural Science Foundation of Fujian (2019J01471) and Quanzhou Program of High-level Talents Innovation and Entrepreneurship (2018C050R).