
Abstract
Three different classes of specific therapy exist for pulmonary arterial hypertension. Ambrisentan belongs to the endothelin receptor antagonist (ERA) class of drugs, which inhibit the action of Endothelin-1; a potent vasoconstrictor and mitogen. Unlike bosentan and sitaxentan, it has a propanoic-acid based structure and like sitaxentan it has selective affinity for type A Endothelin receptors. Two large randomized controlled trials (ARIES-1 and ARIES-2) have demonstrated clinical benefit with ambrisentan in pulmonary arterial hypertension by repeated measurement of 6-minute walk distance. The most common adverse effect associated with ambrisentan use is peripheral edema, the incidence of liver enzyme elevation seen is lower than for other ERAs. Ambrisentan is safe in combination with warfarin and sildenafil. It offers further flexibility in the treatment of PAH as monotherapy and in combination. Although encouraging, trial data do not exhibit improved efficacy compared with other ERAs or sildenafil and there are no direct comparison studies. Long-term outcome data, including subgroup analysis are awaited to see if there are particular benefits of this therapy in PAH.
Introduction
Pulmonary Arterial Hypertension (PAH) is a chronic, debilitating, progressive condition. Over the past ten years there has been much progress in the recognition, understanding and treatment of this important type of pulmonary hypertension. The broader diagnosis of pulmonary hypertension includes all causes of raised pulmonary artery pressure: left sided heart disease, chronic hypoxemia or pulmonary disease, thromboembolic disease and miscellaneous conditions. PAH is the diagnosis applied to those cases that fit into group 1 of the Venice classification of pulmonary hypertension. 1
The gold standard diagnostic test for PAH is a right heart catheter study. Diagnosis requires a mean pulmonary artery pressure (mPAP) of greater than 25 mmHg at rest or 30 mmHg on exercise, a pulmonary vascular resistance (PVR) greater than or equal to 240 dynes/s/cm5 and a pulmonary capillary wedge pressure (PCWP) of less than or equal to 15 mmHg. 1 There are many proponents of using a cut-off value of 20 mmHg at rest for the mean PAP or at least labeling these patients with pre-pulmonary hypertension. The diagnostic criteria and classification were reviewed at the 4th international pulmonary hypertension meeting at Dana Point in 2008 and updated guidelines are scheduled to be published later this year.
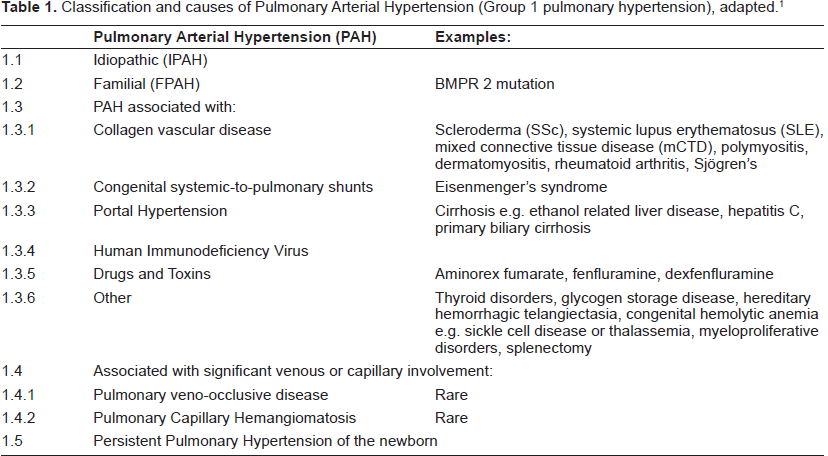
Overall PAH remains a rare disorder that is difficult to detect, frequently it takes months before a diagnosis is made. 2 In untreated cases survival is lower than many cancers and despite advances in treatment mortality remains unacceptably high. 3 There is a wide variety of causes of PAH or group 1 pulmonary hypertension (see Table 1). However, they share many clinical, pathological and therapeutic similarities. PAH occurring in the absence of any known disease associations is termed idiopathic PAH where there is no family history, or familial PAH in the presence of a family history. The commonest underlying genetic abnormality seen in familial PAH is a heterozygous loss of function mutation in the bone morphogenetic protein receptor type II.4,5 The combined incidence of familial and idiopathic is just two persons per million per year and as such they represent a minority of PAH cases but the majority of those enrolled in trials. 6
Connective tissue disease associated PAH (PAH-CTD) is seen in association with: Systemic Scleroderma (SSc—diffuse and limited cutaneous types), systemic lupus erythematosus (SLE), mixed connective tissue disease (mCTD), polymyositis, dermatomyositis, Sjögren's syndrome and rheumatoid arthritis. 7 The incidence of PAH-CTD in the UK is conservatively estimated at 1.27 per million per year. 8 The prevalence of PAH in SSc is reported to be somewhere between 11 and 35%. 2 Annual screening is advocated in limited cutaneous SSc and mCTD patients who are U1 RNP antibody positive due to the frequency of PAH in these groups.9,10 Although improving, prognosis in PAH-CTD historically has been worse than IPAH. There is a paucity of data concerning outcomes in causes of PAH-CTD other than scleroderma (the predominant condition), however SLE patients seem to have a favorable survival based on registry data. 8
There are a large number of congenital lesions with systemic to pulmonary shunts that generate PAH and eventually the Eisenmenger syndrome (reversal of flow through the shunt leading to cyanosis). In the absence of complex lesions outcomes are generally favorable as the right-to-left shunt preserves cardiac output and right ventricle remodeling occurs very gradually. 11 A trial using the endothelin receptor antagonist bosentan in Eisenmenger patients showed significant improvement in exercise capacity and hemodynamics, without adversely affecting arterial saturations. 12
Anorexigen use has produced clusters of PAH cases; the association was discovered in the 1960's when an epidemic occurred in Germany, Austria and Switzerland following the use of aminorex fumarate. 13 More recently fenfluramine and dexfenfluramine have also been implicated. 6 In patients infected with the Human Immunodeficiency Virus (HIV) PAH is rare, with an annual incidence of 0.1%, of course this represents an enormous (thousand fold) increase in relative risk compared with the general population. 14 As a result of improved HIV therapy though most deaths in this group occur as a result of PAH.
Other associated causes of PAH are listed in Table 1). Patients with these disorders are rarely included in clinical trials of PAH therapies, but treatment generally follows that prescribed for idiopathic PAH with additional attention to the underlying condition. Group4pulmonaryhypertension or chronic thromboembolic disease patients may also be considered for targeted PAH therapies: if disease is too distal to be resectable surgically, pulmonary hypertension persists or recurs post-operatively. 15 Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis are rare, but possibly underdiagnosed conditions. The diagnostic problem they present is beyond the discussion of this review as is persistent pulmonary hypertension of the newborn for obvious reasons. Further information about the myriad associations with PAH can be found in review articles.7,16–19
There are several assessments used to monitor the progress of PAH. Right heart catheter hemodynamic variables e.g. mPAP, PVR, right atrial pressure (RAP) and Cardiac Index have been shown to be associated with severity. The modified World Health Organization functional classification (FC) allows for simple, if subjective stratification of patients (Table 2). 19 The most widely used functional measure is the 6-minute walk distance (6 MWD)—which is as straightforward as it sounds—ideally with additional oxygen saturation measurements. Clinical trials infrequently recruit patients with walking distances over 450 m as there may be a treatment ceiling effect. 20 B-type natriuretic peptide (BNP) and the more stable N-terminal cleavage fragment of pro-BNP (NT-pro-BNP) are generated in response to increased ventricular wall tension. In the absence of renal disease or left ventricular impairment, levels and serial changes in BNP or NT-pro-BNP are useful prognostic indicators, which correlate with survival in PAH. 21
Pulmonary function testing and high resolution computed tomography of the chest are both useful to establish the presence of significant lung disease. Echocardiography with Doppler estimation of the pulmonary artery systolic pressure lacks the accuracy and reproducibility to be used for diagnosis. There are studies which use more complex echocardiographic measures to assess progression of PH and response to therapy, but these techniques are not in wide use. 22 Measures used to assess patient's symptoms and quality of life include the visual analogue Borg dyspnea score, the Camphor (Cambridge Pulmonary Hypertension Outcome Review) questionnaire in addition to surveys not specifically designed for the breathless patient e.g. Medical Outcomes Study 36-item Short Form health survey (SF-36). 23
The underlying pathophysiology of PAH is characterized by increasing pulmonary microvascular resistance, due to intimal hyperplasia and smooth muscle hypertrophy of pulmonary arterioles. 7 In severe forms the proliferation of smooth muscle cells generates a concentric neointima. These changes also result in a predilection for in situ thrombosis. The hallmark pathological plexiform lesion results from abnormal endothelial cell proliferation. 24 Vasodilator mechanisms appear to be deficient in PAH with reduced production of endothelial-derived prostacyclin, nitric oxide (NO) synthase and increased thromboxane A2. 25
There are three main targets for therapy in PAH: increasing prostacyclin by using analogues, improving endogenous NO levels by phosphodiesterase-5 inhibition and blocking the receptors and hence the effects of endothelin-1 (ET-1). 7 ET-1 is the dominant isoform of endothelin; a 21 amino acid peptide neurohormone. It is a potent vasoconstrictor that is both produced and secreted by vascular endothelium. There are several stimuli that increase ET-1 levels: hypoxia, ischemia, shear stress, growth factors, angiotensin II and norepinephrine. 26 ET-1 has several other actions: stimulation of smooth muscle cell and fibroblast proliferation, modulation of the sympathetic nervous and renin-angiotensin-aldosterone systems, and pro-inflammatory. ET-1 levels are raised in the lungs and plasma of patients with PAH in a manner that correlates with disease severity and survival. 27
Two different receptors for ET-1 exist in the arterial bed: endothelin receptor type A (ETA) found on smooth muscle cells and fibroblasts and endothelin receptor type B (ETB) present in the endothelium and smooth muscle cells. 7 The effects of ET-1 are dependent upon the receptor activated (see Fig. 1). In normal pulmonary arteries binding to ETA induces vasoconstriction which persists after ET-1 unbinds, whereas ETB stimulation results in transient vasodilatation via NO and prostacyclin. 7 ETB on endothelial cells mediate the clearance of ET-1 and activation also inhibits endothelin converting enzyme-1 which produces mature ET-1. 28 With the increase in ET-1 seen in PAH there is concomitant up-regulation of ETA and ETB receptors, particularly in distal vessels. This increase in ET-1 has also been shown to correlate with mean right atrial pressure, itself a predictor of mortality.29,30 Functional blocking of ET-1 receptors has been shown to reverse vascular remodeling in experimental models. 28
In animal and clinical studies ERA therapy has produced vasodilation and clinical improvement regardless of whether the agent is non-selective or selective. However, the vasodilating effect of ETA blockade is attenuated by concomitant ETB antagonism. 31 The hypothesis that selective blockade of ETA can prevent vasoconstriction and proliferation, whilst allowing ETB mediated vasodilatation and ET-1 clearance, is appealing but the benefit of this approach remains unproven in clinical trials. There are several possible reasons for this. ETB on smooth muscle cells have been observed to form heterodimers with ETA and mediate vasoconstriction. 32 Selective antagonism may lead to compensation by other receptor subtypes, so-called ‘cross-talk’. 33 In the PAH disease state a sub-population of ETB on fibroblasts and smooth muscle cells may effect vasoconstriction, proliferation and fibrosis. 28
Mechanism of Action, Metabolism and Pharmacokinetic Profile
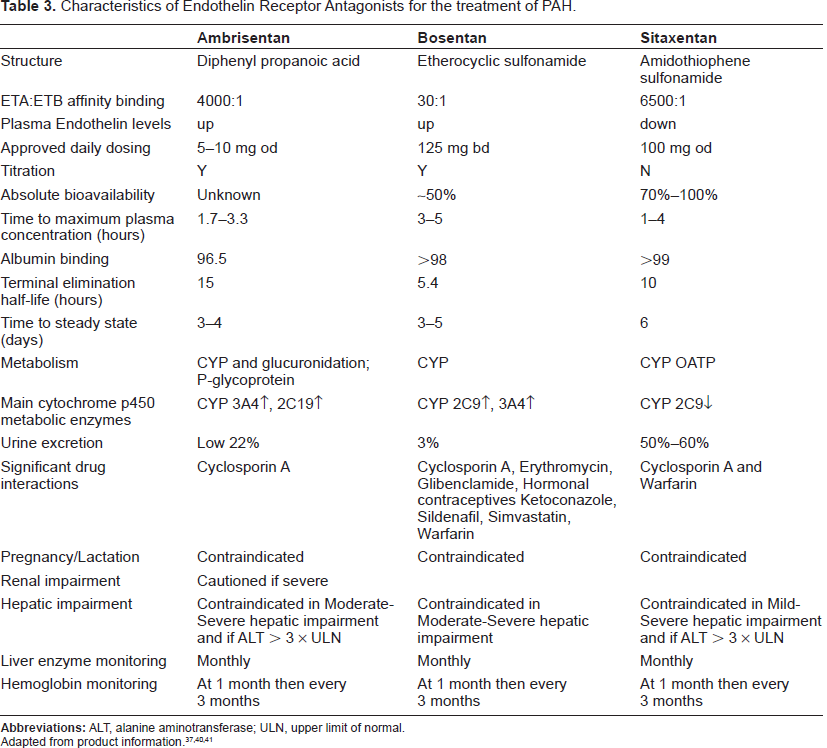
Ambrisentan, chemical formula of C22H22N2O4, is considered, like sitaxentan, to be selective for ETA, unlike bosentan, which is referred to as a dual endothelin receptor antagonist. The generally held belief is that molecules are ETA selective if they exhibit >100:1 affinity as assessed by in vitro competitive receptor assays. 34 By this method ambrisentan may only have an affinity of 77:1 compared with 20:1 for bosentan and 6500:1 for sitaxentan. 34 However, various values are quoted for the relative affinity for ETA to ETB for most ERAs. One study using myocardial membranes reported ambrisentan to have an affinity of > 4000:1 compared to only 23:1 for bosentan and 900:1 for sitaxentan. 35 Experiments using the basilar arteries of rats concluded that ETB inhibition meant ETA affinity must be lower than 77:1. 36 Although more reproducible, in vitro assays do not necessarily concur with in vivo studies which have looked at functional selectivity; by measuring ET-1 levels. Less selective ERAs, including ambrisentan and bosentan increase ET-1 levels two hours after ingestion, whereas a reduction has been observed after administration of sitaxentan. 28 The questions; what is the best way to assess selectivity? and what properties make a molecule selective? remain unanswered. Until clinical data suggest that the difference is significant in treating human disease a distinction is not entirely helpful to the clinician. It would be reasonable to conclude, relative to its competitors, ambrisentan is more selective than bosentan, but less selective than sitaxentan for ETA.
Ambrisentan differs from bosentan and sitaxentan by virtue of having a propanoic acid side-chain rather than a sulfonamide group. The main mechanism of metabolism is glucuronidation (via UGT1A9S, UGT2B7S and UGT1A3S) with some contribution from oxidative metabolism (CYP3A4 mainly, also CYP3A5 and CYP2C19). 37 In animal models ambrisentan did not significantly inhibit or induce hepatic enzymes even beyond therapeutic levels, this may be beneficial in preventing liver toxicity. 2 Elimination is mostly via the biliary system with 22% of oral doses recovered in the urine. 37
In clinical trials the pharmacokinetics of ambrisentan were studied with a maximum plasma concentration at steady state ranging from 111 to 1,223 ng/ml occurring 1.7 to 3.3 hours after dosing. 38 Dose-linear pharmacokinetics are observed between doses of 1 and 100 mg. Steady state takes 4 days with once daily dosing. 37 Ambrisentan is 99% protein bound with an elimination half-life of 9-15 hours. 39 There is no available data concerning ambrisentan use in severe renal impairment or haemodialysis but in those with a creatinine clearance greater than 20 ml/min no dose adjustment is required. Despite structural differences with other ERAs hepatic impairment would still be expected to affect pharmacokinetics and therefore ambrisentan is not recommended in moderate to severe hepatic impairment. As a class of drugs ERAs are considered teratogenic, and are not recommended for lactating women either. Table 3 summarizes and compares the pharmacological properties of the three ERAs in current use.
Potential interactions of ambrisentan have been evaluated with warfarin and sildenafil; two frequently used therapies in PAH. Prothrombin time did not significantly alter in healthy volunteers given warfarin before and after an eight day course of ambrisentan. 42 Warfarin use was permitted in the ARIES trials and those results concurred with observations that no dose adjustment is necessary. 43 A crossover study was used to establish that ambrisentan and sildenafil can be safely used together—no significant change in the pharmacokinetics of either drug was found. 44
Ambrisentan is a substrate of P-glycoprotein and organic anion transport protein both of which are inhibited by cyclosporine A so, as for other ERAs, co-administration is contraindicated. Many of the interactions of ambrisentan have not been characterized but caution when using CYP3A4 inhibitors e.g. clarithromycin, itraconazole and protease inhibitors, CYP2C19 inhibitors e.g. omeprazole or CYP inducers e.g. rifampicin continues to be advisable. 45
Clinical Studies
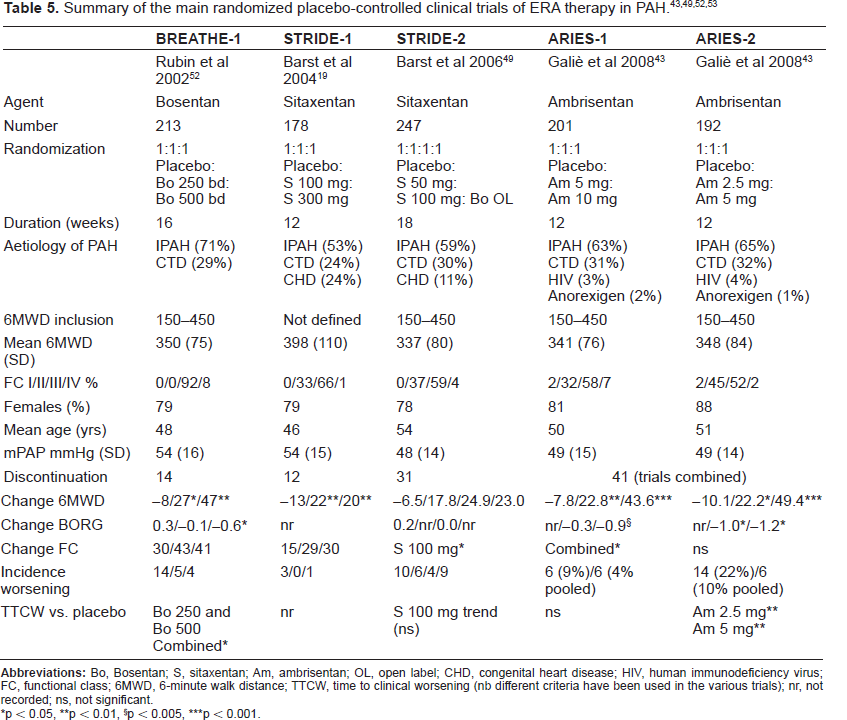
In 2005, Galiè et al published the results of a dose-ranging study using ambrisentan. Patients (n = 64) with IPAH (39), PAH-CTD (19), anorexigen use PAH (4) or HIV-PAH (2) in functional classes II (23) or III (41) were recruited for the 24 week study. 38 Exclusion criteria included 6 MWD of < 150 m or > 450 m, chronic use of prostanoid or ERA therapy within the 4 weeks prior to study entry and serum aminotransferase levels >1.5 times the upper limit of normal. The dose-ranging double-blind phase of the study was conducted for 12 weeks and followed by a further 12 week open-label extension period. Participants were randomized to once daily doses of 1, 2.5, 5 or 10 mg of ambrisentan, those in the 5 and 10 mg dose groups were up-titrated from an initial dose of 2.5 mg. The protocol allowed a reduction or cessation of the study drug if aminotransferase levels became elevated (>3 or >5 times the upper limit of normal) according to treating physician during the open-label phase. 44 The primary endpoint was change in 6 MWD at 12 weeks, with secondary endpoints of change in: Borg dyspnea index, WHO functional class, global assessment of quality of life and time to clinical worsening. Of the patients remaining on ambrisentan, 48% had been up-titrated to 10 mg once daily dosing by the end of the study.
The Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES-1 and ARIES-2) were phase 3 trials conducted concurrently in different parts of the world. 43 Patients with PAH were stratified as either IPAH or other causes (PAH-CTD, HIV-associated or anorexigen use associated PAH were included) were assigned to receive placebo, 5 or 10 mg ambrisentan daily (ARIES 1) and placebo, 2.5 or 5 mg (ARIES-2). Exclusion criteria included 6 MWD < 150 or > 450 and the current use of PAH specific therapies.
The primary endpoint was the change in 6 MWD from baseline to 12 weeks with secondary measures of change in WHO functional class, change in health survey score, time to clinical worsening (TTCW), Borg dyspnea score and plasma BNP concentration. 49 Clinical worsening events included were: death, lung transplantation, hospitalization for PAH, atrial septostomy, study withdrawal because of the addition of other PAH medications, or early escape criteria. Although published in one paper the studies underwent separate statistical analysis with a descriptive comparison of the combined placebo and 5 mg treatment groups. Beyond 12 weeks an open label extension was conducted in which placebo patients also received ambrisentan. 43 For the analysis patients who received a single dose of ambrisentan were included. Where data was missing at 12 weeks baseline values were imputed unless a patient discontinued because of clinical worsening (in which case the worst possible score was used e.g. FC IV, 6 MWD 0 m). At baseline overall 38% of patients were in FC II and 55% in FC III with a predominance of IPAH patients. 43 The open label follow up extension study ARIES-E, allowed further dose increases after 24 weeks and data has been analyzed at 48 weeks. 46
Safety
The ARIES trials included early escape criteria which permitted premature study discontinuation if within four weeks there were two of the following: greater than 20% reduction in 6 MWD, increase in WHO functional class, worsening right ventricular failure, progressive hepatic or renal failure, systolic blood pressure <85 mmHg. In total there were sixteen instances of early escape, only five were from treatment arms. 43 The absolute number of patients deteriorating was less on ambrisentan therapy than placebo: in ARIES-1 there were two deaths in the placebo group and one each in the treatment arms, in ARIES-2 three patients died whilst taking placebo, two died whilst taking 2.5 mg and none on the 5 mg dose. 43 Overall there were six discontinuations (2.3%) because of adverse events (worsening PAH, worsening dyspnea, gastroenteritis, intracranial bleeding, allergic reaction, headache/face edema), in the patients receiving ambrisentan in ARIES. 43
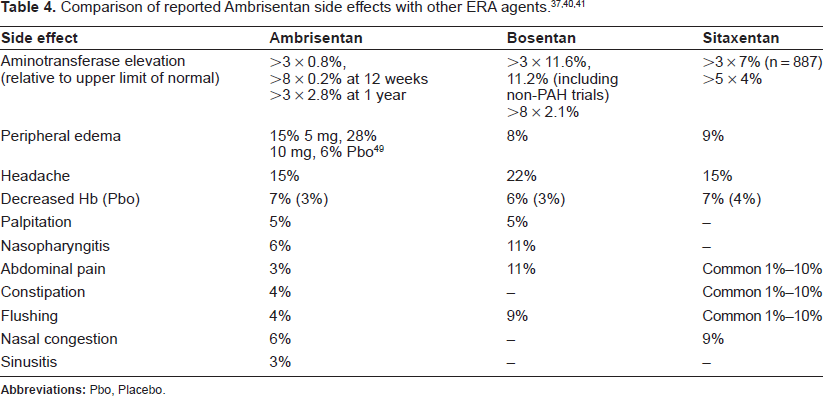
In the initial study of ambrisentan six patients did not complete the 12 week study, three of these were because of serious events: two sudden deaths and one instance of aminotransferase elevation. 38 A further patient (also on 5 mg dose) developed aminotransferase elevation requiring therapy cessation. 38 They also reported elevation of >3 times upper limit of normal (ULN) in a patient on anti-retroviral therapy. Two patients on the 2.5 mg dose had elevation >3 times ULN which spontaneously resolved on repeat testing without a change in treatment. 38 In the extended study no further increases in aminotransferase levels >3 times ULN occurred. 47 There were no reported elevations of hepatic enzymes >3 times ULN in ARIES-1 and ARIES-2 trials, but eight such instances reported in extended follow up.43,46
Additionally, a small study has been conducted in patients who had previously failed on ERA therapy due to liver enzyme elevation. Thirty-six patients were enrolled, who previously discontinued bosentan (n = 31), sitaxentan (n = 2) or both (n = 3); at baseline 69.4% were receiving prostanoid or sildenafil. With up-titration from 2.5 mg to 5 mg after 4 weeks, then to 10 mg in over half the patients and a median exposure of 102 weeks there was only one incidence of liver enzyme elevation >3 times ULN which resolved after dose reduction. 48 The small number of patients with previous exposure to sitaxentan in this trial makes it difficult to be sure that ambrisentan will be safe, with respect to liver toxicity, in such cases. ERA-induced liver toxicity has been noted to occur many after many months on therapy and progression to fulminant hepatitis and death has been seen with sitaxentan. 49 The mechanism of bosentan toxicity has been studied in animal models suggesting potential explanations of impaired bile salt transport via the canalicular bile salt export pump. 50
Hemoglobin levels have been noted to fall with ERA therapy, usually within a few weeks of commencing therapy. In the ARIES trials hemoglobin concentration fell by 0.8 g/dl at 12 weeks compared with a slight increase of 0.2 g/dl seen in the placebo groups. 43 In keeping with other observations no further fall occurred in the following 12 week open label phase. 38 Ambrisentan is not recommended for patients with significant anemia and monitoring of the full blood count every three months is advised.
The most frequently reported side effect with ambrisentan is peripheral edema with an incidence of 17-25% across all doses.38,43 Edema occurs more frequently and with greater severity in patients over the age of 65 and also when using the 10 mg dose. 37 As alluded to in the trial reports, edema frequently requires an escalation of diuretic therapy. Peripheral edema is seen in approximately 10% of ERA trial patients treated with placebo, in which case it may reflect deterioration of right heart failure. This does not exclude ERA exacerbating this problem in the treatment arms. A straightforward explanation for the edema is that in common with other side effects it is due to small vessel vasodilatation. An alternative possibility is ETB blockade impairing regulation of epithelial sodium channels in the renal tubules. 34 Side effects reported in the ARIES trials include (combined rate for all treatment groups): headache 18%, nasopharyngitis 9%, nasal congestion 5%, palpitations 5%, flushing 4%, constipation 4%, sinusitis 3%, and abdominal pain 3%. 43 In the earlier study nasal congestion, upper respiratory tract infection, headache, flushing and nausea were commonly reported but not in a dose-related manner. 38 Table 4 shows the comparative incidence of the more common side effects seen with ERA therapy.
Efficacy
The main efficacy endpoint tested in trials of ambrisentan is improvement in 6 MWD. With doses of 1 mg, 2.5 mg, 5 mg and 10 mg 6 MWD improved significantly at 12 weeks. 38 Whilst data for the IPAH subgroup were suggestive of a dose-response relationship, the overall data did not support this conclusion. 38 The twenty-four week data from the study extension showed a better 6 MWD response for the IPAH group compared with the other causes of PAH (+64.3 compared with + 38.5 m, p < 0.05 two-sample t-test). 47 ARIES showed a placebo-corrected increase in 6 MWD in each dose group at week 12 compared with placebo of; 31 m (p = 0.008) and 59 m (p < 0.001) for 5 and 10 mg respectively in ARIES-1, 32 m and 59 m for 2.5 mg and 5 mg respectively in ARIES-2 (p < 0.001). 43 6 MWD in the combined 5 mg dose group improved by 45 m with a p-value of < 0.001. 43 Integrated data from 383 patients on ambrisentan monotherapy showed that improvement in 6 MWD had been maintained at 48 weeks (36 m) from 38 m at twelve weeks. 46
Time to clinical worsening was shown to be longer with ambrisentan (log-rank test, p = 0.005) in the combined 5 mg dose group and in ARIES-2 at both the 2.5 and 5 mg (p < 0.01) doses, but not in ARIES-1. 43 In both ARIES trials clinical worsening was defined by the combined end points of death, lung transplantation, hospitalization for PAH, atrial septostomy, study withdrawal due to escape criteria or study withdrawal due to the addition of other PAH therapeutic agents. The extended ARIES-E data reports an event free survival of 79.2% for CTD-PAH and 85.5% for IPAH after one year. 51 In the 2005 Galiè ambrisentan trial clinical worsening criteria included an increase in the patient's diuretic requirement. Of the thirteen and eight patients reported to have deteriorated from 0-12 and 12-24 weeks respectively, eleven of them were due to increases in diuretic therapy. 38 Survival at 12 weeks in patients treated with ambrisentan in clinical trials was 97%, 99% and 98% in the 2005 study, ARIES-1 and ARIES-2 respectively.38,43 In the 48 week extension of the ARIES trials survival was 95%. 51 The ARIES-E data in abstract form reported a 1 year estimated survival of 90.3% in the 124 enrolled CTD-PAH patients. 51
Borg dyspnea scores at 12 weeks compared with placebo improved significantly for the 10 mg dose and the combined 5 mg and 10 mg data in ARIES-1 and the 2.5 mg and 5 mg doses separately and combined in ARIES-2 and the combined 5 mg dose group. 43 In the earlier study improvements seen in Borg dyspnea index at 12 weeks were maintained at 24 weeks in all doses (p < 0.0001 compared with baseline). 38 In ARIES-1 the number of subjects whose functional class deteriorated at 12 weeks was less with ambrisentan than placebo resulting in a significant improvement in functional class distribution. A similar trend seen in ARIES-2 was not significant, but the combined 5 mg data also showed significant improvement. 43 In the Galiè et al 2005 study the functional class improvement seen in a few patients at 12 weeks was maintained to 24 weeks. 47
BNP data from 110 patients in ARIES-1 showed both the 5 mg and 10 mg dose reduced BNP levels compared with placebo (p < 0.005 and p < 0.001). In ARIES-2 a similar effect was seen (p < 0.005 for both 2.5 mg and 5 mg dose); data from 107 patients was available. 43 The ARIES trials did not include repeat assessment of hemodynamic variables. Of the 64 patients in the dose-ranging study there is only follow up right heart catheter data for 29 patients. Ambrisentan would appear to produce a good hemodynamic response with a significant fall in mPAP and PVR. 38 This is comparable to other ERA therapies in trials although the lack of corroborative data and the small number of patients suggest further work would be beneficial. Table 5 summarizes the results of the principal clinical trials of ERA therapy.
Patient Preference
A visual analogue scale from 0 (very poor) to 100 mm (excellent) was used to determine improved subject wellbeing with ambrisentan. The baseline score was improved by 11.3 mm at week twelve for all dose groups combined, this was sustained at 24 weeks (+12.1 mm). 38 Even with a penalty value of 0 imputed for missing data at 12 weeks the improvement was significant. There were three voluntary withdrawals from therapy, one in the randomized phase and two during the open label extension. Six patients withdrew from the ARIES trials. The SF-36 health survey results in ARIES were inconsistent (the cultural differences between the different regions of the world may be a significant factor). ARIES-1 data only suggests trends. ARIES-2 patients reported significant improvement compared with placebo. This applied to the physical functioning scale in both dose groups. Improvements were also suggested in the role physical, vitality, role-emotional and general health scores. 43
Once daily dosing of ambrisentan is particularly likely to be appealing for those patients taking multiple medications. Experience suggests that for many patients a monthly blood test for liver enzyme levels is an unwanted burden. If necessary, this can be avoided by treating with sildenafil. Given the widespread use of warfarin in PAH with regular INR monitoring, in practice the required liver tests infrequently require additional blood samples.
The high prevalence of peripheral edema has led to a FDA warning and from the trial this side effect seems to be the biggest difficulty with ambrisentan. Fortunately it is a relatively simple problem to correct with diuretics. By taking preemptive action patient compliance may be improved as a negative experience is averted. Before starting or increasing ambrisentan, commencing or increasing diuretic therapy should be considered, particularly if there is mild edema present. For those patients intolerant of ERA therapy due to adverse symptoms there is no evidence to suggest they will cope any better with ambrisentan; a trial period on low dose may be adopted for such individuals.
Place in Therapy
Ambrisentan therapy has been shown to be effective in PAH. It was granted a license for the treatment of PAH in patients in FCII and III in the USA in 2007 and subsequently other countries have followed suit. A recent meta-analysis showed treatments for PAH are effective at improving mortality, 6 MWD, functional class status and hemodynamic variables and reducing hospital admissions. 54 Included within this meta-analysis were results of trials using oral sildenafil, intravenous epoprostenol, inhaled iloprost and subcutaneous trepostinil.58–68 At present there is little data showing direct comparison between the available therapies and choice of therapy may be dependent on wider issues. As further research and trials into the treatment of PAH continue, it might be hoped that the optimal treatment strategy will become clear.
At present several questions remain unanswered e.g. the theoretical advantage of selective ETA blockade—this has not been borne out in the clinical trials conducted thus far. Indeed, there is currently a dearth of data comparing the oral therapies (including sildenafil) used in PAH. Comparable efficacy data from the clinical trials of ambrisentan, bosentan and sitaxentan are limited to the 6 MWD and do not clearly favor either therapy. The greater experience with bosentan and the relative wealth of long-term safety and survival data make it hard to preferentially recommend any therapy before it on grounds of effectiveness. In cases where bosentan therapy has been stopped due to side effects (particularly liver enzyme elevation) ambrisentan is a suitable alternative or adjunct if sildenafil has been started.
Another issue gaining attention is the use of combination therapy. American registry data (REVEAL) reports over a quarter of PAH patients to be on two or more drugs targeting different therapeutic pathways. 66 Whilst synergistic effects might be hoped for, any additional benefit may be a victim to the law of diminishing returns and unwanted effects may also be increased. The BREATHE-2 trial added placebo or bosentan at commencement of treatment with iv epoprostenol, but only showed a trend towards benefit in the bosentan arm. 62 The PACES study showed a significant improvement in 6 MWD and TTCW when sildenafil was added to iv epoprostenol. 56 Inhaled iloprost has been shown to have an additive effect when added to oral therapy. 63 The EARLY study allowed patients on stable doses of sildenafil to be enrolled and subgroup analysis showed a significant reduction in PVR in those already on sildenafil. 67 In the recently concluded PHIRST study, tadalafil (a phosphodiesterase-5 inhibitor) was used in naïve patients or those already on bosentan. Although the treatment effect was greater in the treatment naïve group, the bosentan subgroup exhibited an improved 6 MWD. 68 Many combinations of drugs have not been assessed specifically including those utilizing the selective ERA antagonists.
The majority of data from clinical trials concern patients with IPAH and the ARIES trial data strongly support the use of ambrisentan. Subgroup analysis showed greater improvement for IPAH patients than CTD-PAH (ranging from 50 to 66 m and 15 to 23 m respectively. 43 This does not translate to a recommendation of ambrisentan in preference to other oral therapies. ARIES included a significant minority of CTD-PAH patients. There is suggestive evidence that selective ETA blockade may improve outcomes in CTD-PAH from studies comparing sitaxentan and bosentan. 49 Ambrisentan is not as selective as Sitaxentan but might be expected to achieve similar results. The available data from ARIES-E shows a trend towards improved 6 MWD and functional class for the 5 mg and 10 mg dose. 51 The change appears to be more marked at 24 weeks than 12 or 48 weeks but in keeping with other trials any treatment effect is smaller than that seen in the IPAH group. Perhaps a better indication of efficacy is the event free survival with ambrisentan estimated at 79.2% for CTD-PAH (versus 85.6% in IPAH); this compares favorably with historical data. 51
The BREATHE-5 trial showed bosentan improved exercise capacity, without reducing peripheral oxygen saturation in PAH associated with congenital systemic-to-pulmonary shunts. 12 It remains to be seen if ambrisentan is effective as this group was not included in the ARIES trial. PAH associated with HIV, although represented in ARIES there were too few patients to make subgroup analysis meaningful. In PAH associated with drugs, congenital hemolytic anemia and in chronic thromboembolic disease clinical data is too sparse to make recommendations but guidance generally follows treatment for IPAH with consideration of the individual and concomitant therapy.
Patients in WHO functional class II were well represented in ARIES (38% of total patients) and exhibited a similar improvement (range, 36 to 55 m) to those in WHO functional class III (range, 39 to 45 m). 43 ARIES-E reports an improvement in 6 MWD of 61.7 m at 48 weeks in the 104 patients who were functional class II at baseline and received ambrisentan. 69 The increase in 6 MWD seen in class II patients treated with bosentan in the EARLY study at 6 months was 19.1 m. 67
The apparent advantage to ambrisentan over the other ERAs is the lower incidence of hepatic enzyme elevation, but these rates may not be at equipotent doses. Analysing the data, there remains sufficient doubt concerning the safety of ambrisentan to refrain from using it in patients with abnormal aminotransferases (>1.5 times the upper limit of normal) or moderate hepatic impairment. Whilst ambrisentan is a better choice than bosentan or sitaxentan in the presence of significant liver disease e.g. porto-pulmonary hypertension, sildenafil is a safer oral option. The overall incidence of liver enzyme elevation quoted in the trials is low, but an annual incidence of 2.8% still requires monthly monitoring which can be difficult for some patients. It has been shown to be safe if there has been prior aminotransferase elevation with bosentan treatment and does not affect warfarin levels.
Conclusions
Ambrisentan is an ERA (endothelin-1 receptor antagonist), which shows preferential affinity for the type A endothelin-1 receptor (ETA) over the type B receptor (ETB). It has been shown in two large, randomized-placebo controlled trials to be effective in PAH at improving exercise tolerance as measured by the 6 MWD. Additional secondary measures of improvement including time to clinical worsening, survival, functional class, quality of life and hemodynamic variables have been reported in trials. A favorably low incidence of aminotransferase elevation indicating hepatic toxicity has been observed compared with sulfonamide based ERAs. It can be safely administered with warfarin or sildenafil without need for dose adjustment of either therapy. A once daily oral medication with relatively few side effects is an attractive option; especially as the use of therapies in combination continues to increase. Long-term data regarding survival and disease progression are awaited before the relative clinical benefits can be compared with other ERAs. Ambrisentan provides another valuable, effective treatment option in PAH.
Disclosure
The authors report no conflicts of interest.
Footnotes
Appendix 1
Appendix 2
Classification and causes of Pulmonary Arterial Hypertension (Group 1 pulmonary hypertension), adapted.1
| Pulmonary Arterial Hypertension (PAH) | Examples: | |
|---|---|---|
| 1.1 | Idiopathic (IPAH) | |
| 1.2 | Familial (FPAH) | BMPR 2 mutation |
| 1.3 | PAH associated with: | |
| 1.3.1 | Collagen vascular disease | Scleroderma (SSc), systemic lupus erythematosus (SLE), mixed connective tissue disease (mCTD), polymyositis, dermatomyositis, rheumatoid arthritis, Sjögren's |
| 1.3.2 | Congenital systemic-to-pulmonary shunts | Eisenmenger's syndrome |
| 1.3.3 | Portal Hypertension | Cirrhosis e.g. ethanol related liver disease, hepatitis C, primary biliary cirrhosis |
| 1.3.4 | Human Immunodeficiency Virus | |
| 1.3.5 | Drugs and Toxins | Aminorex fumarate, fenfluramine, dexfenfluramine |
| 1.3.6 | Other | Thyroid disorders, glycogen storage disease, hereditary hemorrhagic telangiectasia, congenital hemolytic anemia e.g. sickle cell disease or thalassemia, myeloproliferative disorders, splenectomy |
| 1.4 | Associated with significant venous or capillary involvement: | |
| 1.4.1 | Pulmonary veno-occlusive disease | Rare |
| 1.4.2 | Pulmonary Capillary Hemangiomatosis | Rare |
| 1.5 | Persistent Pulmonary Hypertension of the newborn |
Appendix 3
Functional Classification of Pulmonary Hypertension modified after the NYHA functional classification according to the World Health Organization. 19

| Class I—Patients with pulmonary hypertension but without resulting limitation of physical activity. Normal physical exertion does not cause undue dyspnea or fatigue, chest pain or near syncope. |
| Class II—Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope. |
| Class III—Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary physical activity causes undue dyspnea or fatigue, chest pain or near syncope. |
| Class IV—Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity. |
Appendix 4
Characteristics of Endothelin Receptor Antagonists for the treatment of PAH.
| Ambrisentan | Bosentan | Sitaxentan | |
|---|---|---|---|
| Structure | Diphenyl propanoic acid | Etherocyclic sulfonamide | Amidothiophene sulfonamide |
| ETA:ETB affinity binding | 4000:1 | 30:1 | 6500:1 |
| Plasma Endothelin levels | up | up | down |
| Approved daily dosing | 5-10 mg od | 125 mg bd | 100 mg od |
| Titration | Y | Y | N |
| Absolute bioavailability | Unknown | ~50% | 70%-100% |
| Time to maximum plasma concentration (hours) | 1.7-3.3 | 3-5 | 1-4 |
| Albumin binding | 96.5 | >98 | >99 |
| Terminal elimination half-life (hours) | 15 | 5.4 | 10 |
| Time to steady state (days) | 3-4 | 3-5 | 6 |
| Metabolism | CYP and glucuronidation; P-glycoprotein | CYP | CYP OATP |
| Main cytochrome p450 metabolic enzymes | CYP 3A4↑, 2C19↑ | CYP 2C9↑, 3A4↑ | CYP 2C9↓ |
| Urine excretion | Low 22% | 3% | 50%-60% |
| Significant drug interactions | Cyclosporin A | Cyclosporin A, Erythromycin, Glibenclamide, Hormonal contraceptives Ketoconazole, Sildenafil, Simvastatin, Warfarin | Cyclosporin A and Warfarin |
| Pregnancy/Lactation | Contraindicated | Contraindicated | Contraindicated |
| Renal impairment | Cautioned if severe | ||
| Hepatic impairment | Contraindicated in Moderate-Severe hepatic impairment and if ALT > 3 × ULN | Contraindicated in Moderate-Severe hepatic impairment | Contraindicated in Mild-Severe hepatic impairment and if ALT > 3 × ULN |
| Liver enzyme monitoring | Monthly | Monthly | Monthly |
| Hemoglobin monitoring | At 1 month then every 3 months | At 1 month then every 3 months | At 1 month then every 3 months |
Appendix 5
Comparison of reported Ambrisentan side effects with other ERA agents.37,40,41
| Side effect | Ambrisentan | Bosentan | Sitaxentan |
|---|---|---|---|
| Aminotransferase elevation (relative to upper limit of normal) | >3 × 0.8%, >8 × 0.2% at 12 weeks >3 × 2.8% at 1 year | >3 × 11.6%, 11.2% (including non-PAH trials) >8 × 2.1% | >3 × 7% (n = 887) >5 × 4% |
| Peripheral edema | 15% 5 mg, 28% 10 mg, 6% Pbo 49 | 8% | 9% |
| Headache | 15% | 22% | 15% |
| Decreased Hb (Pbo) | 7% (3%) | 6% (3%) | 7% (4%) |
| Palpitation | 5% | 5% | – |
| Nasopharyngitis | 6% | 11% | – |
| Abdominal pain | 3% | 11% | Common 1%-10% |
| Constipation | 4% | – | Common 1%-10% |
| Flushing | 4% | 9% | Common 1%-10% |
| Nasal congestion | 6% | – | 9% |
| Sinusitis | 3% | – | – |
Appendix 6
Summary of the main randomized placebo-controlled clinical trials of ERA therapy in PAH.43,49,52,53
| BREATHE-1 | STRIDE-1 | STRIDE-2 | ARIES-1 | ARIES-2 | |
|---|---|---|---|---|---|
| Rubin et al 2002 52 | Barst et al 2004 19 | Barst et al 2006 49 | Galiè et al 20 08 43 | Galiè et al 2008 43 | |
| Agent | Bosentan | Sitaxentan | Sitaxentan | Ambrisentan | Ambrisentan |
| Number | 213 | 178 | 247 | 201 | 192 |
| Randomization | 1:1:1 | 1:1:1 | 1:1:1:1 | 1:1:1 | 1:1:1 |
| Placebo: | Placebo: | Placebo: | Placebo: | Placebo: | |
| Bo 250 bd: | S 100 mg: | S 50 mg: | Am 5 mg: | Am 2.5 mg: | |
| Bo 500 bd | S 300 mg | S 100 mg: Bo OL | Am 10 mg | Am 5 mg | |
| Duration (weeks) | 16 | 12 | 18 | 12 | 12 |
| Aetiology of PAH | IPAH (71%) | IPAH (53%) | IPAH (59%) | IPAH (63%) | IPAH (65%) |
| CTD (29%) | CTD (24%) | CTD (30%) | CTD (31%) | CTD (32%) | |
| CHD (24%) | CHD (11%) | HIV (3%) | HIV (4%) | ||
| Anorexigen (2%) | Anorexigen (1%) | ||||
| 6MWD inclusion | 150-450 | Not defined | 150-450 | 150-450 | 150-450 |
| Mean 6MWD | 350 (75) | 398 (110) | 337 (80) | 341 (76) | 348 (84) |
| (SD) | |||||
| FC I/II/III/IV % | 0/0/92/8 | 0/33/66/1 | 0/37/59/4 | 2/32/58/7 | 2/45/52/2 |
| Females (%) | 79 | 79 | 78 | 81 | 88 |
| Mean age (yrs) | 48 | 46 | 54 | 50 | 51 |
| mPAP mmHg (SD) | 54 (16) | 54 (15) | 48 (14) | 49 (15) | 49 (14) |
| Discontinuation | 14 | 12 | 31 | 41 (trials combined) | |
| Change 6MWD | -8/27*/47** | -13/22**/20** | -6.5/17.8/24.9/23.0 | -7.8/22.8**/43.6*** | -10.1/22.2749.4*** |
| Change BORG | 0.3/-0.1/-0.6* | nr | 0.2/nr/0.0/nr | nr/-0.3/-0.9$d§ | nr/-1.0*/-1.2* |
| Change FC | 30/43/41 | 15/29/30 | S 100 mg* | Combined* | ns |
| Incidence worsening | 14/5/4 | 3/0/1 | 10/6/4/9 | 6 (9%)/6 (4% pooled) | 14 (22%)/6 (10% pooled) |
| TTCW vs. placebo | Bo 250 and Bo 500 Combined* | nr | S 100 mg trend (ns) | ns | Am 2.5 mg** Am 5 mg** |
p < 0.05,
p < 0.01, §p < 0.005,
p < 0.001.