
Abstract
In recent years in the management of pulmonary arterial hypertension (PAH), endothelin receptor antagonists (ERAs) represent a well-established class of therapeutic agents with clear beneficial effects. Macitentan (Opsumit®), a dual ERA optimized for efficacy and safety, is the newest drug in the class. Macitentan presents a number of key beneficial characteristics, including increased in vivo preclinical efficacy versus existing ERAs, resulting from sustained receptor binding and physicochemical properties that allow enhanced tissue penetration. The clinical pharmacokinetics studies also indicated a low predilection of macitentan for drug–drug interactions. In the SERAPHIN trial, a phase III long-term study of PAH, macitentan significantly reduced morbidity and mortality by 45% versus placebo, providing sustained long-term improvements in exercise capacity. No association was found between changes in exercise capacity and long-term clinical outcomes, but improved cardiopulmonary hemodynamics were recorded in macitentan-treated patients irrespective of baseline background PAH therapy or World Health Organization functional class. Based on these favorable data, the US Food and Drug Administration approved the 10 mg/day dose in late 2013 and the same process has recently been concluded by the European Medicines Agency.
Introduction
If untreated, pulmonary arterial hypertension (PAH) is a progressive and fatal disease characterized by elevated pulmonary arterial pressure that leads to right ventricular failure. The classification for pulmonary hypertension contains 5 groups. PAH is listed in group 1 and the forms included are idiopathic, heritable or associated with: connective tissue disease, congenital heart disease, HIV infection, portal hypertension, and exposure to toxins/drugs (Simonneau et al., 2009). PAH treatments have transformed the prognosis for PAH patients from symptomatic improvements in exercise tolerance to delayed disease progression and even reversibility in selected forms. Improved disease awareness and evidence-based guidelines developed from randomized, controlled clinical trial data have highlighted the need for early intervention, goal-oriented treatment and combination therapy [Galiè et al. 2009]. Research from the past 20 years has helped to identify three major pathways of endothelial dysfunction implicated in the pathogenesis of PAH. For each of the prostacyclin, nitric oxide and endothelin pathways, various molecules have been synthesized in order to correct endothelial dysfunction that ultimately have been translated into clinical improvements.
The endothelin pathway
Endothelin 1 (ET-1), a highly active peptide, was found to be implicated in one of the main physiopathological pathways leading to intense vascular remodeling of the small pulmonary arteries and thus responsible for the development of PAH. It is a very potent vasoconstrictor, with a proliferative effect on vascular smooth muscle cells as it interacts with two types of receptors: Endothelin receptor A (ETA) is localized mainly on the pulmonary artery smooth muscle cell and ETB is localized mainly on the vascular endothelium [Raja and Dreyfus, 2008; Raja, 2010]. ETA isoform activation together with activation of ETB within the smooth muscle cell leads to vasoconstriction and proliferation of vascular smooth muscle cells [Pollock, 2005]. ETB receptors also play an important role in clearance of ET-1 (as a scavenger receptor), particularly in the vascular beds of the lungs and kidneys by removing ET-1 from the circulation [Dupuis et al. 1996]. ETB receptors may also be responsible for nitric oxide and prostacyclin release from the endothelial cells leading to vasodilatation in the above mentioned areas (Figure 1) [Hirata et al. 1993; Rubin et al. 2005].

Schematic representation of the endothelin system in vascular tissue.
This is probably why the immediate response to an intravenous bolus of ET-1 is a decrease in vascular resistance, which is primarily mediated by the release of nitric oxide and prostacyclin from ETB-stimulated endothelial cells [Berti et al. 1993; Filep et al. 1995]. Subsequently, the increase in vascular resistance (that may be sustained for up to 1 h) is mediated via ETA-stimulation of pulmonary artery smooth-muscle cells (PASMC) (Wenzel et al. 1998). Despite the opposing effects of the activated ETA and ETB receptor, it is generally accepted that, under pathological conditions, high levels of ET-1 result in vasoconstriction [Ortega Mateo and de Artiñano, 1997]. Plasma and lung expression of ET-1 is increased in patients with PAH and correlates with disease severity, thus playing a central role in the imbalance between vasodilatation and vasoconstriction specific to this disease [Giaid et al. 1993; Rubens et al. 2001]. Recent results indicate that in general both ETA and ETB receptors are reduced in PAH, specifically ETB, and that ETB signaling through protein kinases is markedly reduced in PASMC from PAH patients carrying a BMPR 2 mutation, while remaining stable in idiopathic PAH [Yu et al. 2013].On the other hand, further data suggest that in states of inflammation, increased ETB-receptor expression and activation mediated by elevated ET concentrations may be an underestimated mechanism [Juergens et al. 2008].
In preclinical models of PAH, blocking the endothelin system with endothelin receptor antagonists (ERAs) can improve hemodynamics, right ventricular hypertrophy and survival [Dupuis and Hoeper, 2008; Dupuis and Prié, 1999; Prié et al. 1997, 1998]. After extended research both selective and nonselective ERAs are now approved and available for treating PAH. It has been shown that nonselective ERAs increase plasma ET-1 via blockade of ETB receptors, which are responsible for the clearance of ET-1 at the level of the lung [Fukuroda et al. 1994].
There are currently two ERAs available on the market for treating PAH: bosentan and ambrisentan. Bosentan (Tracleer, Actelion, Switzerland) is a dual ET receptor blocker with similar affinity for ETA and ETB receptors [Dupuis and Hoeper, 2008], available since the beginning of the last decade, with a moderate liver toxicity. The latest guidelines recommend it for patients in the World Health Organization functional classes (WHO FC) II, III and IV [Galiè et al. 2009, 2013]. Ambrisentan (Volibris, GlaxoSmithKline, UK) is an oral selective ETA receptor antagonist which, although favoring fluid retention, has a lower rate of hepatic injury [Galiè et al. 2013]. It is also recommended for patients in WHO FC II, III and IV [Galiè et al. 2009]. A third previously available selective ETA called sitaxentan was withdrawn from the market in 2010 due to an increasing number of deaths attributed to acute liver failure [Lavelle et al. 2009].
Macitentan
Macitentan, also called Actelion-1 or ACT-064992, is a new oral dual (ETA and ETB) ERA which has recently been developed for the treatment of PAH. It was developed by Actelion, its chemical formula being N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (Figure 2). It was synthesized from the potassium salt of propyl sulfamide and 5-(4-bromophenyl)-4,6-dichloro-pyrimidine [Bolli et al. 2012]. Much has been done in recent years to study the drug’s pharmacokinetics and pharmacodynamics.

Chemical formula of macitentan.
Pharmacokinetics
Absorption
Macitentan presents a 40- and 2000-fold increased affinity for the lipid phase versus bosentan and ambrisentan [Iglarz et al. 2008]. It has a slow absorption – a maximum plasma concentration (Cmax) of 170 ng/ml at 6 h after dosing with a 5–10 h range and geometric mean terminal halflife (T1/2) of 15 h. For ACT-132577, which is macitentan’s major metabolite, Cmax (121 ng/ml) was reached 48 h after dosing and its T1/2 was 44 h [Bruderer et al. 2012b].
Metabolism
Macitentan’s metabolic pathway, and the overall elimination of the drug, is catalyzed by the cytochrome P450 (CYP) system, especially CYP3A4 and to a lesser extent CYP2C19 isoenzyme. Macitentan is not a substrate of P-glycoprotein. Thus, hepatic uptake takes place mostly by passive diffusion, being independent of organic anion-transporting polypeptide transport [Atsmon et al. 2013].
Following administration of a single oral 10 mg dose of 14C-macitentan to healthy subjects, results showed that macitentan undergoes two major metabolic reactions: oxidative depropylation and oxidative cleavage. ACT-132577 is formed by oxidative depropylation and subsequently undergoes conjugation with glucose to form M706u, a metabolite which is eliminated in urine but is not present in feces. ACT-132577 is a major metabolite, reaching plasma exposure levels four to five times higher than macitentan; it is also a dual ERA [Bolli et al. 2012; Iglarz et al. 2008; Bruderer et al. 2012b]. Oxidative cleavage of the ethylene glycol linker helps form the carboxylic acid, ACT-373898, which in turn undergoes hydrolysis to form M323u. Both ACT-373898 and M323u are present in urine and feces, and constitute the major radiolabeled products in urine. Mean (± standard deviation) cumulative recovery of radioactivity from feces and urine was 73.6% (±6.2%) of the administered radioactive dose, with 49.7% (±3.9%) cumulative recovery from urine and 23.9% (±4.8%) from feces. Thus urinary excretion was a more important route of elimination than feces in humans. ACT-373898 and ACT-132577 represented approximately 20% and 190%, respectively, of the radioactivity of macitentan and about 8% and 60%, respectively, of the total plasma radioactivity in the studied plasma pools. In urine, four entities were identified, none of them being macitentan or ACT-132577. In feces, five entities were identified, with ACT-080803, the hydrolysis product of macitentan (-37.7%) and ACT-132577 (-14.0%) being the most abundant ones [Bruderer et al. 2012b].
Concentration: plasma, blood
Following the same 10 mg administration of 14C-macitentan, measured concentrations of total radioactivity in whole blood were lower than in plasma. Cmax and area under the curve from zero to time t [AUC(0-t)] values of total radioactivity in plasma were approximately 80% and 96%, respectively, greater than in whole blood with an AUC(0-∞) difference of 105%, which shows that macitentan and its metabolites bind poorly to or penetrate red blood cells/erythrocytes [Bruderer et al. 2012b].
At a dose of 300 mg the pharmacokinetics of macitentan are less than dose-proportional; ACT-132577 plasma concentration of the metabolite was higher and elimination was slower. This resulted in a total exposure to the metabolite that was 2.7-fold [95% confidence interval CI) 2.5–2.9] greater than that of macitentan at a dose of 600 mg. Macitentan dose-dependently increased ET-1 concentrations up to 2.2-fold (95% CI 1.4–2.4) at a dose of 600 mg, but had no consistent effect on total bile salts [Sidharta et al. 2011]. Cmax for macitentan and ACT-132577 exceeded the IC50 value measured in vitro, confirming the pharmacological effect of ACT-132577 [Iglarz et al. 2008].
In subjects with known mild, moderate or severe hepatic impairment who received macitentan 10 mg per day, no relevant clinical differences were found in pharmacokinetics compared with healthy subjects after a survey of 21 days. Plasma concentrations of macitentan and ACT-132577 were generally lower in subjects with hepatic impairment than in healthy subjects, but no correlation was found between the severity of hepatic impairment and plasma concentrations of macitentan or its metabolites [Sidharta et al. 2012].
Pharmacodynamics
In vitro effects
Macitentan administration causes a dose-dependent increase in plasma ET-1 with maximum effects (a 2-fold increase) attained at 10 mg [Sidharta et al. 2013]. Concerning direct tissue effects macitentan and ACT-132577, both inhibit ET-1 induced contraction in rat isolated tissue preparations (aorta denuded of endothelium; ETA receptor-mediated) and also inhibit sarafotoxin S6c-mediated contraction of rat trachea denuded of epithelium (ETB receptor-mediated) by inducing a parallel shift to the right in the concentration response curve for ET-1 mediated contraction, but with no significant change in the maximum response to ET-1 or sarafotoxin S6c [Iglarz et al. 2008].
Both macitentan and ACT-132577 are dual receptor antagonists. Compared with macitentan, ACT-132577 is approximately 5-fold less potent on ETA receptors and presents an ETA/ETB inhibitory potency ratio of 16 versus 50 for its parent compound, further adding to the pharmacological effect of the parent compound [Iglarz et al. 2008]. Further in vitro assays showed that macitentan and ACT-132577 fully antagonized intracellular calcium increase induced by ET-1 on nonrecombinant cells (PASMC and rat aortic smooth muscle cell line A10 and mouse fibroblast cell line 3T3) [Iglarz et al. 2008].
In PASMC, dissociation kinetics and receptor occupancy half-life (ROT1/2) of macitentan, ambrisentan and bosentan were examined. Results showed a marked difference in the ROT1/2 of macitentan, which demonstrated a 15-fold increase compared with ambrisentan and bosentan (ROT1/2: 17 minutes versus 40 seconds and 70 seconds, respectively) (Gatfield et al. 2012). Induced vessel contraction essays by exogenously added ET-1 showed an increased potency of macitentan compared with bosentan or ambrisentan regarding the inhibition of intracellular calcium response to ET-1 stimulation in PASMC. Furthermore, during the same phase of the experiment, macitentan showed no surmountable behavior to the ET-1-induced signal of the PASMC as opposed to bosentan and ambrisentan. The effect was seen only in short ET-1 stimulation times. Thus in PASMC, macitentan, but not ambrisentan and bosentan, displayed slow receptor dissociation causing insurmountable antagonism in ET-1-induced inositol-1-phosphate (IP1) accumulation and ET-1-induced sustained elevation of intracellular calcium levels [Gatfield et al. 2012].
In vivo effects
Macitentan has a potent pharmacological activity in vivo. When administered to normotensive rats, macitentan increased plasma ET-1 concentration, thus confirming its dual ETA/ETB receptor antagonism. This increase occurred at a 10-fold lower dose than with bosentan. In hypertensive rats, macitentan and bosentan decreased mean arterial blood pressure with macitentan having a more potent effect. At the maximal effective dose, the duration of the blood pressure response to macitentan was 2-fold longer than bosentan (40h versus 20 h) [Iglarz et al. 2008]. The blood pressure lowering effect proved to be also dose-dependent in hypertensive rats [Bolli et al. 2012] and also in humans when compared with enalapril [Clozel, 2006].
Macitentan in PAH
The SERAPHIN study
The safety and efficacy of macitentan in PAH patients was evaluated in the SERAPHIN study (Study with an Endothelin Receptor Antagonist in Pulmonary arterial Hypertension to Improve clinical outcome). This was the first and largest phase III long-term, multicenter, double-blind, placebo-controlled, event-driven study in PAH. It was a large study including 742 patients and stretched out over a long median treatment period which exceeded 2 years. SERAPHIN study has a unique and original design, being an event-driven outcome trial. This means that the study was pursued until it achieved certain predefined goals. It is also macitentan’s single published randomized, controlled trial to date.
Methods
A total of 742 patients from 151 centers in 39 countries diagnosed with PAH were enrolled worldwide. The repartition for each group was as follows: idiopathic n = 404 (55%); heritable n = 13 (1.8%); related to connective-tissue disease n = 224 (30.5%); repaired congenital systemic-to-pulmonary shunts n = 62 (8.4%); HIV infection n = 10 (1.4%); and associated with drug use or toxin exposure n =22 (3%). The patients eligible for inclusion in the trial were randomized to receive two different doses of macitentan (3 and 10 mg once daily) or placebo with approximately 250 patients in each of the three groups. Patients could have been on other concomitant PAH treatment throughout the study (oral phosphodiesterase type 5 inhibitors, oral or inhaled prostanoids, calcium channel blockers or l-arginine) without changes within the last 3 months, while patients receiving intravenous or subcutaneous prostanoids were excluded. The mean duration of study treatment was 85.3 weeks for the patients receiving placebo, 99.5 weeks for the 3 mg/day dose of macitentan and 103.9 weeks for the 10 mg/day dose of macitentan (Figure 3).
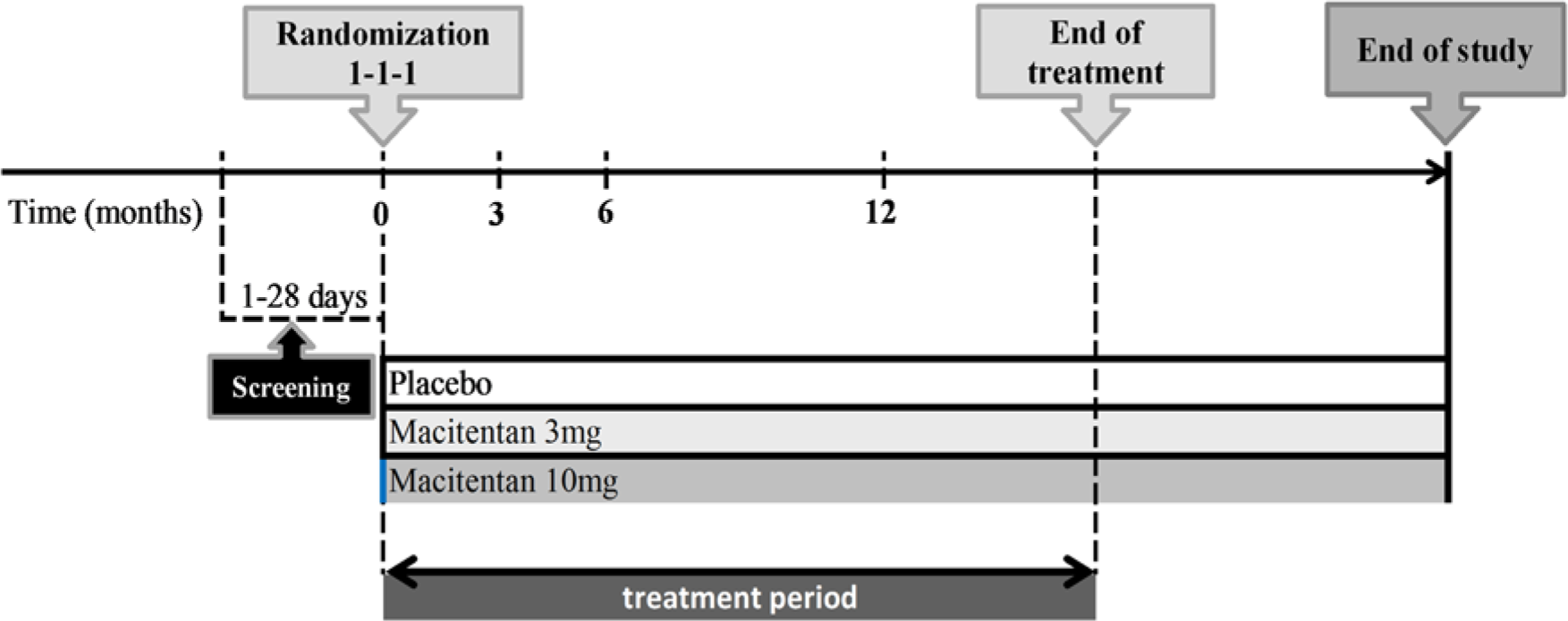
SERAPHIN study design.
The composite primary endpoint was the time from the initiation of treatment to the first event related to PAH. Such events were defined as worsening of PAH, initiation of treatment with intravenous or subcutaneous prostanoids, lung transplantation, atrial septostomy and death from any cause up to the end of treatment. Worsening of PAH was described as a decrease in the 6-minute walk distance (6MWD) of at least 15% from baseline together with the worsening of symptoms of PAH, namely ‘change from baseline to a higher WHO FC, or no change in patients who were in WHO FC IV at baseline, and the appearance or worsening signs of right heart failure that did not respond to oral diuretic therapy and that prompted for additional treatment for PAH’ Pulido et al. [2013]. Secondary endpoints included: change from baseline to month 6 in the 6MWD; percentage of patients with an improvement in WHO FC at month 6; death due to PAH or hospitalization for PAH up to the end of treatment; and death from any cause up to the end of treatment and up to the end of the study.
Results
The results showed that the primary endpoint occurred in 46.4% (116 patients) in the placebo group, 38% (95 patients) in the macitentan 3 mg/day and 31.4% (76 patients) in the macitentan 10 mg/day group (Table 1). The most frequent primary endpoint event was worsening of PAH with a hazard ratio (HR) for macitentan 3 mg/day versus placebo of 0.70 (97.5% CI, 0.52–0.96; p = 0.0108) and the HR for macitentan 10 mg/day versus placebo being 0.55 (97.5% CI, 0.39–0.76; p < 0.0001). The composite endpoint of death due to PAH or hospitalization for PAH occurred in 84 patients (33.6%), 65 patients (26.0%), 50 patients (20.7%) in the placebo, 3 mg/day of macitentan and 10 mg/day of macitentan groups, respectively, with a HR of 0.67 (97.5% CI, 0.46–0.97; p = 0.01) for the 3 mg/day dose of macitentan versus placebo and 0.50 (97.5% CI, 0.34–0.75; p < 0.001) for the 10 mg/day dose of macitentan versus placebo. After the same 6 months interval the WHO FC improved in 13% of patients in the placebo group, 20% in the 3 mg/day (p = 0.04) and 22% (p = 0.006) in the 10 mg/day dosage of macitentan.
Primary and secondary endpoints for events related to pulmonary arterial hypertension and death. (Adapted from Pulido et al. [2013].).
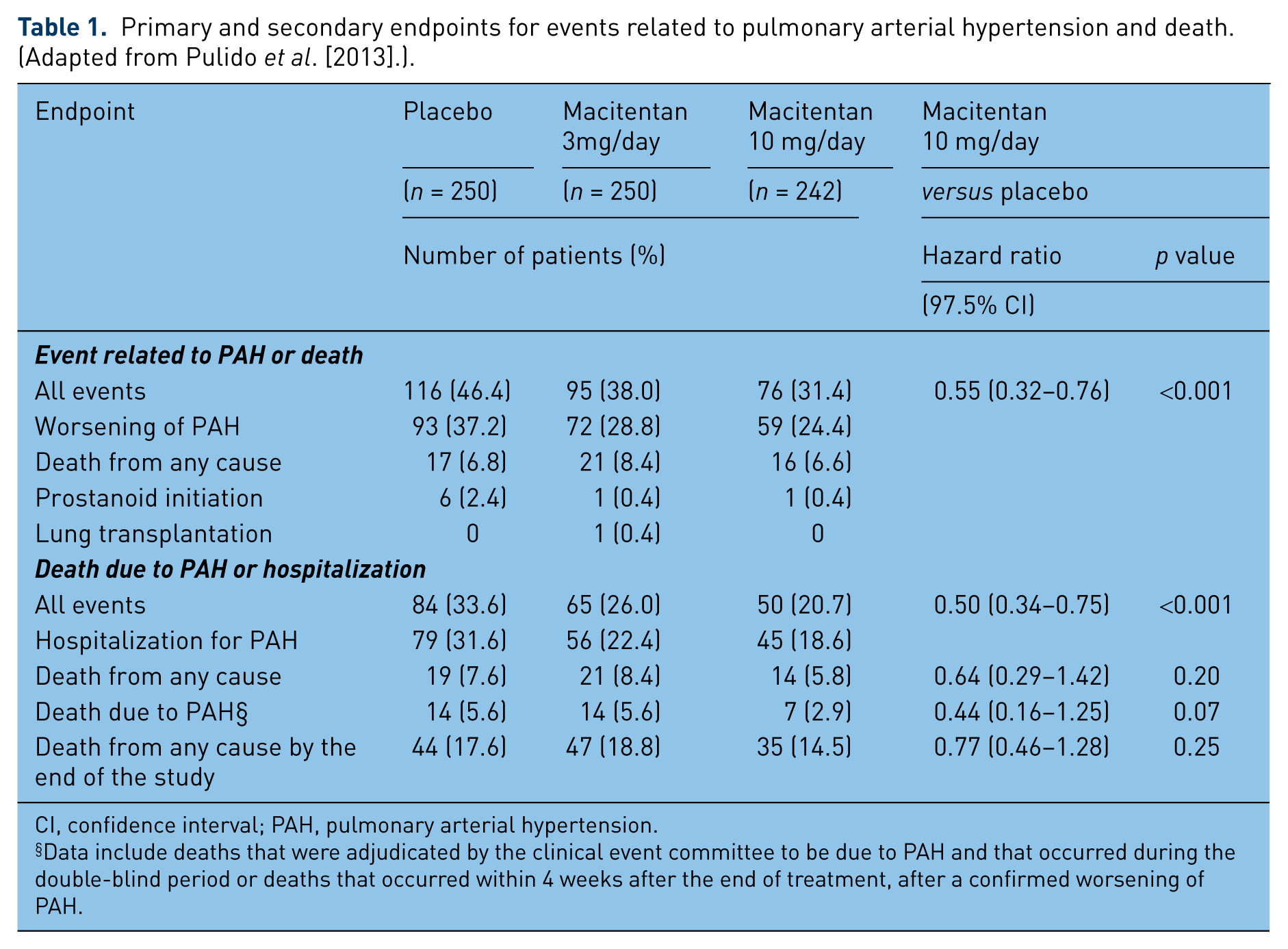
CI, confidence interval; PAH, pulmonary arterial hypertension.
Data include deaths that were adjudicated by the clinical event committee to be due to PAH and that occurred during the double-blind period or deaths that occurred within 4 weeks after the end of treatment, after a confirmed worsening of PAH.
Results showed that after 3.5 years of treatment, macitentan (in both 3 and 10 mg daily regimens) successfully reduced the risk of a morbidity/mortality event over the treatment period versus placebo in a dose-dependent manner: a 45% reduction (p < 0.0001) in the 10 mg/day group and a 30% reduction (p = 0.01) in the 3 mg/day group, respectively. There were also improvements in the 6MWD, with an increase for the 3 mg/day dose regimen by a mean of 7.4 months (treatment effect versus placebo of 16.8 months; 97.5% CI, −2.7 to 36.4; p = 0.01) and by a mean of 12.5 months (treatment effect versus placebo of 22.0 months; 97.5% CI, 3.2–40.8; p < 0.01) for the 10mg dose regimen. Improvements in the WHO functional class were also recorded [Pulido et al. 2013]. These secondary efficacy endpoints showed a dose-dependent effect with p < 0.05 for both dose regimens.
Further studies in the Eisenmenger syndrome (MAESTRO) study and Digital Ulcers treatment in Systemic Sclerosis Patients (DUAL-2) study are currently underway to determine a possible beneficial effect of this new ERA.
Toxicity
In the SERAPHIN trial, macitentan 3 and 10 mg /day was well tolerated with the numbers of adverse events and patients discontinuing treatment due to side effects being similar in both groups. The incidence of serious adverse events was higher in patients treated with placebo compared with macitentan, with 55% of patients in the placebo group experiencing serious adverse events compared with 52% and 45% of patients in the macitentan 3 mg and 10 mg/day groups, respectively. Compared with placebo, a higher proportion of macitentan-treated patients had nasopharyngitis, headache and anemia. The number of patients with elevated liver alanine or aspartate aminotransferases higher than 3 times the upper limit of normal were similar in all groups (4.5% of patients receiving placebo, 3.6% of patients on 3 mg/day macitentan and 3.4% of patients on 10 mg /day macitentan). Another reported side effect was the decrease in hemoglobin (<10 g/dl), more frequently encountered in the macitentan group (6.2% of patients on 3 mg/day and 8.7% of patients on 10 mg/day) than in the placebo group (3.4%). Once detected, it showed no difference after treatment discontinuation between the two groups, with one patient in each treatment group discontinuing treatment due to anemia. Furthermore, no difference in fluid retention/edema between groups was noted with an incidence of 16–18, 2–18 and 1% within the macitentan 3 mg/day, 10 mg/day and the placebo groups, respectively.
Liver toxicity appears to be an effect of the ERA drug class [Battistini et al. 2006]. However, macitentan requires a lower dose compared with bosentan in order to be efficient in the PAH phenotype. Thus it is possible that macitentan could be associated with reduced liver toxicity compared with the other ERAs [Pollock et al. 2009]. In contrast, it is more likely to increase plasma ET-1 concentrations compared with other selective or dual ET antagonists [Pollock et al. 2009].
In other aforementioned human trials, dose levels of up to 300 mg (maximal tolerated dose) were well tolerated and the frequency of adverse events was similar to that in the placebo group. Headache, nausea and vomiting were dose-limiting adverse events with no consistent effect on total bile salts [Sidharta et al. 2011]. Other human trials reported no effect on bile salts with the most frequently reported side effect being headache [Sidharta et al. 2013; Bruderer et al. 2012a]. In addition, concomitant administration of cyclosporine, rifampicin or ketoconazole did not appear to modify tolerability of macitentan [Bruderer et al. 2012a; Atsmon et al. 2013]. However, concomitant treatment with cyclosporin did not have any clinically relevant effect on the exposure to macitentan or its metabolites at steady-state whereas concomitant treatment with rifampicin reduced significantly the exposure to macitentan [Bruderer et al. 2012a]. Macitentan administration also caused a small increase in 6β-hydroxycortisol/cortisol but the increase was not statistically significant for any dose [Sidharta et al. 2013]. At a dose of 10 mg once daily, blood pressure, heart rate, clinical laboratory, electrocardiographic parameters and vital signs did not record significant clinical modifications [Kummer et al. 2009; Bruderer et al. 2013].
Thus, current recommendations state that macitentan should not to be initiated in patients with severe hepatic impairment or elevated aminotransferases (>3 times normal) and it is not recommended in patients with moderate hepatic impairment. After treatment onset, monthly monitoring of alanine aminotransferase (ALT) and aspartate transaminase (AST) is advised. If clinically relevant aminotransferase elevations occur, or if elevations are accompanied by an increase in bilirubin >2 times normal, or by clinical symptoms of liver injury (e.g. jaundice), treatment should be discontinued. Regarding hemoglobin concentration, current recommendations do not advise treatment initiation in patients with severe anemia. It is recommended that hemoglobin concentrations be measured prior to treatment onset and tests repeated during treatment as clinically indicated.
Conclusion
Up until the 1990s the only treatment for PAH was pulmonary transplantation. The survival rate in PAH patients has improved dramatically in the past 20 years with the development of specific PAH treatments. Unfortunately the disease remains incurable and frequent adverse effects called for new, more efficient molecules. In this clinical context, macitentan represents a new potent ETA/ETB receptor antagonist, compatible with once-a-day dosing regimens. The SERAPHIN study confirmed macitentan’s efficacy in PAH by significantly reducing the risk of morbidity and mortality by 45% and also the risk of PAH-related death or hospitalization by 50% versus placebo. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMEA) approved macitentan for the treatment of PAH.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Andrei Seferian, David Montani, Laurent Savale, Xavier Jaïs, Florence Parent, Marc Humbert Olivier Sitbon, Gerald Simonneau, have relationships with drug companies including Actelion, Bayer, GSK, Novartis, and Pfizer. In addition to being investigator in trials involving these companies, relationships include consultancy service and membership of scientific advisory boards.