
Abstract
Lennox-Gastaut syndrome is an epileptic encephalopathy starting in early childhood consisting of the triad of cognitive impairment, multiple seizures types and slow spike-wave complexes in the electroencephalogram. Global developmental delay is usually evident before the onset in patients with symptomatic LGS whereas children with cryptogenic LGS might have a normal history of cognitive development before onset of the seizures. The seizure types most commonly associated with LGS are tonic seizures, myoclonic, dialeptic (absence) and astatic seizures. The seizures usually respond poorly to antiepileptic drugs. Cognitive deterioration occurs even in individuals who developed normally before onset of the encephalopathy. In this review, we will focus on the medical, dietary and surgical treatment of seizures associated with this syndrome. The treatment options for LGS have gained recently by emerging randomized controlled trials of new antiepileptic drugs and the introduction of vagal nerve stimulation.
Keywords
Introduction
Epileptic encephalopathies are disorders in which the epilepsy by itself is thought to contribute significantly to mental deterioration and loss of intellectual abilities. In general, these syndromes are believed to be age related reactions to non-specific and divers exogenous insults to the developing brain. 1 Examples of age-specific encephalopathies are Othahara syndrome, Dravet syndrome, West syndrome and Lennox-Gastaut syndrome (LGS). LGS is a severe epileptic encephalopathy usually affecting preschool-aged children but might develop as early as at one year or as late as at eight years of age. The syndrome was established in 1966 by the Marseille School in France considering the work of Dravet, Sorel and Gastaut and co-workers. 2 It was adopted by the International League Against Epilepsy Classification Commission in 1989. 3 It is defined by the electroclinical hallmarks of drug resistant seizures, mental deterioration and an EEG pattern consisting of generalized slow spike-waves complexes. 4 The most common seizures types are tonic, dialeptic (absence), myoclonic and astatic seizures. In addition, generalized tonic-clonic and simple and complex-motor seizures might occur. 5 Status epilepticus might appear as stuporuos states (atypical absence status) or be associated with recurrent myoclonic and tonic seizures. Seizure frequency is usually high and daily seizures are common. The majority of patients (up to 80%) have an underlying neurological disease. These patients define the “symptomatic” group of LGS. 6 The remaining 20% of the patients occur without any symptomatic cause and are classified as “cryptogenic” cases. A history of global developmental delay is often present at the onset of symptomatic LGS. In contrast, patients might have had a normal development until clinical onset in the cryptogenic group. 7 Response to medical treatment is poor in the majority of cases. 8 However, recent double-blind, placebo controlled trials suggest a relevant reduction in seizure frequency for some AEDs. 9 The long-term prognosis is poor; although seizures may improve, complete seizure freedom is rare and the mental and psychiatric disorders tend to worsen with time. 10
Epidemiology
LGS accounts for up 10% of cases of epilepsy in the first 5 years of life with little differences among countries in the western world. 11 The annual incidence was reported to be 2 per 100,000 and the prevalence ranges from 0.1 to 0.3 per 1,000 individuals 12 with a mean age of onset between 26 and 28 months of life. 13 However, the interpretation of epidemiological data is hampered by the fact that LGS might be often poorly defined and used for different severe epilepsy syndromes of childhood characterized by multiple types of intractable seizures.
Etiology
LGS is divided into symptomatic or cryptogenic cases by the presence or absence of an underlying neurological disease or brain pathology. The cryptogenic cases have no identifiable underlying cause and show normal findings in neuroimaging studies. They account for about one fourth to one third of all patients with LGS. 13 Symptomatic forms account for the majority of cases of LGS. In these patients, an underlying cause is evident and significant abnormalities might be seen in neuroimaging. Cerebral lesions are especially associated with severe gray matter damage. 8 Common etiologies of symptomatic forms of LGS are summarized the Table 1.
Common etiologies of LGS.

Clinical Presentation
The classical electroclinical triad of LGS includes seizures, mental retardation and an interictal EEG pattern consisting of generalized slow spike-wave complexes and background slowing when the syndrome is completely developed. However, at the onset of the syndrome the full electroclinical picture might not yet been fully established. An underlying neurological disorder might help to identify children with symptomatic LGS. In addition, a history of prior infantile spasms with hypsarrhythmia is reported in approximately 10% to 25% of all patients with LGS.8,13,14
Seizure types
The typical seizure types in LGS include tonic, dialeptic (absence), 5 myoclonic and atonic seizures. In addition, several other less common seizure types occur.
Brief
Several other seizure types arising from focal brain regions such as
A tendency to
Mental development
Cognitive impairment is another typical feature of LGS. The majority of patients exhibit abnormal developmental milestones before the seizures begin. This is especially true for the symptomatic group. Neuroimaging studies reveal significant abnormalities in most of these patients. In cryptogenic cases, the development of the child appears to be normal before the onset of the first seizures. 7 However, cognitive impairment is obvious in 75%–95% of the patients after a mean period of 5 years from onset of the syndrome 20 suggesting that epileptic activity plays a substantial role in the development of cognitive and behavioral deterioration. 9
EEG Features
Interictal EEG
The characteristic interictal EEG pattern of LGS are generalized slow-spike-wave-complexes (SSWC) which repeat at a frequency of 1.5 to 2.5 Hz. They have a frontal or less commonly occipital maximum (Fig. 1). Generalized polyspikes (Fig. 2) and paroxysms of generalized fast rhythmic activity at 10 to 25 Hz (Fig. 3) occur more frequently during sleep. Multiregional spikes and sharp waves may predominate in the EEG 21 Background slowing is commonly seen and correlates with the severity of cognitive impairment. 22
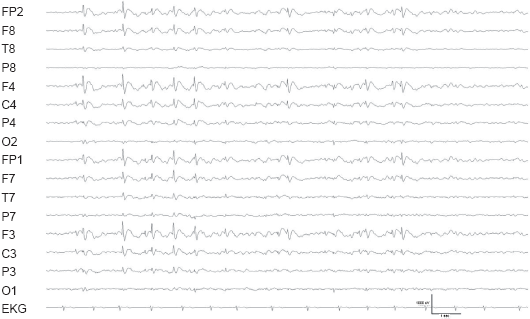
Generalized slow spike-wave complexes in a patient with LGS.
Referential recordings to the the right mastoid electrode.
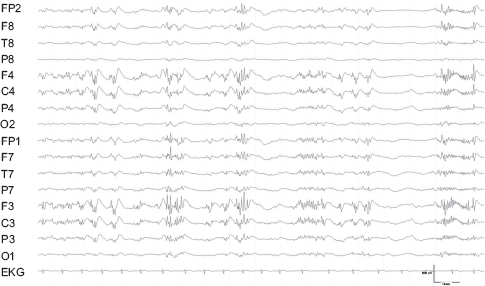
Generalized slow spike-wave complexes with polyspikes preceding the generalized slow spike-wave complexes.
Referential recordings to the the right mastoid electrode.
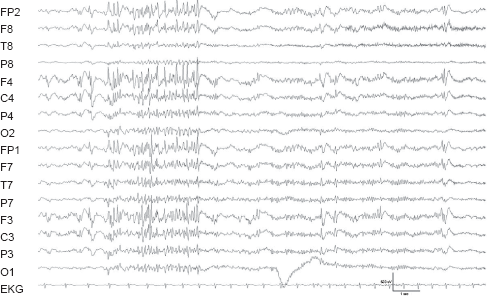
Paroxysms of generalized fast rhythmic activity occurring in sleep in a patient with LGS. *Referential recordings to the the right mastoid electrode.
Ictal EEG
The ictal EEG in LGS is very characteristic. Dialeptic (atypical absence) seizures are associated with generalized SSWC. This pattern is the predominant EEG finding in patients with LGS and it may be difficult to be differentiated by burst of interictal SSWC from ictal activity because the cognitive status of severely retarded children is often difficult to assess. Tonic seizures are typically associated with generalized paroxysmal fast activity and polyspikes in the EEG. In atonic seizures which are associated with reduced electromyographic artifacts in the EEG, a generalized sharp wave might precede a generalized attenuation of EEG activity with low amplitude high frequency activity (Fig. 4).
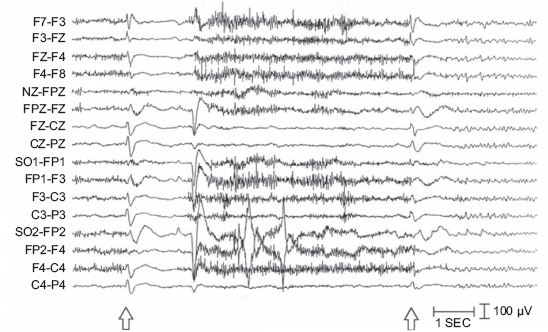
Adult patient with LGS and tonic seizure. Each seizure began with a generalized sharp wave followed by attenuation of EEG activity and cessation of muscle artifact. *Adapted from. 68
Treatment
Medical therapy
One of the hallmarks of the syndrome is medically intractability with poor response of the various seizures types to common AEDs. The aim of medical treatment is to prevent seizures which might harm the patients by causing falls i.e. drop attacks and to minimize the number of daytime seizures to allow the child to attend school or other daily activities. In addition, long lasting periods of prolonged seizures and periods of reduced states of consciousness resembling non-convulsive status epilepticus should be prevented. As frequently combinations of two or three AEDs are needed to establish an acceptable seizure reduction, pharmacokinetics and interactions of the drugs must be considered. Medical treatment of LGS was based on clinical experience and empiric data for decades. In recent years AEDs were investigated in randomized placebo controlled trials which will be discussed in the next paragraphs.
Current Medical Therapy of LGS
Valproic acid is considered the standard first line drug of choice for patients with LGS as it exhibits potential effects against all seizures types which are encountered in LGS despite the fact that it has not been evaluated in randomized clinical trials in LGS yet. Valproic acid has been shown to be effective in children and adolescents with a variety of generalized seizure types by reducing the seizure frequency. In addition, the number of concomitant AEDs can be reduced in some patients. It is the drug of choice for generalized epilepsies and is effective in myoclonic and dialeptic seizures associated with generalized spike-wave discharges. 23 Adverse events which might be encountered are either anorexia in young children or weight gain in older children or adolescents. More severe but rare side effect of valproic acid are idiosyncratic liver failure, acute pancreatitis and thrombocytopenia. 24
Benzodiazepines are also considered as a first line drug. 25 From the three most commonly used benzodiazepines clonazepame, nitrazepame and clobazame, the latter harbors the least sedative effect and might be used preferably. The use of benzodiazepines can be limited by significant sedation or even more commonly by development of tolerance. 26 Benzodiazepines might be tried alone or in combination with other drugs i.e. Valproic acid.
Levetiracetam is an antiepileptic drug with a novel mechanism of action by modulation of synaptic neurotransmitter release through binding to the synaptic vesicle protein SV2A in the brain. Its pharmacokinetic advantages include rapid and almost complete absorption, minimal binding to plasma proteins, absence of enzyme induction, absence of interactions with other drugs, and partial metabolism outside the liver. It has been demonstrated to be effective as adjunctive therapy for refractory partial-onset seizures, generalized tonic-clonic seizures, and myoclonic seizures of juvenile myoclonic epilepsy. 27 Its main adverse effects have been somnolence, asthenia, infection, and dizziness in adults. In children, behavioral adverse effects of hostility and nervousness were also noted. 28 Levetiracetam has not been investigated extensively in LGS patients but the observation that it might reduces myoclonic and tonic-clonic seizures may justify using it in a second line approach. 29
Vigabatrine has been investigated in several trials in patients with various epilepsy syndromes including LGS. Whereas in some of these trials seizure reduction was observed in a considerable portion of individuals, in other trial it was not30,31 making a final conclusion for the use difficult. In addition, the complication of irreversible visual field defects restricted the use of vigabatrine considerably in recent years. 32
Zonisamide is used as an adjunctive therapy for refractory partial seizures with or without secondary generalization in the U.S. and the EU and as a monotherapy in Japan. In general, it is not recommended for use in patients under 18 years of age. A long-term post-marketing survey involving 1631 patients in Japan indicated that 28% of those with West syndrome or LGS improved after treatment with zonisamide. 33
Corticosteroids and intravenous immunoglobulins may also be tried in patients with LGS. It is commonly accepted that the use of these medications have a short time beneficial effect on seizure frequency and the long term results appear to be less promising. 34 In addition, no general recommendations are available regarding the preparation, the dose and the time intervals of these agents. Corticosteroids may lead to severe adverse events when used over longer time periods, thus, limiting its use to short term treatment. Intravenous immunoglobulins are restricted to patients in whom other treatment regimes have failed because of its intravenous application and high costs. 9
Carbamazepine, phenobarbital, phenytoin and ethosuximide are widely used to treat different seizures types of various epilepsy syndromes. However, none of these AEDs is considered as a first or second line drugs as their effect is often restricted to some of the seizures types occurring in LGS. Furthermore, carbamazepine may facilitate dialeptic (absence) seizures especially in children. 35
Pyridoxine is suggested by some authorities to be used as a first line treatment as the syndrome of vitamin B6 dependent seizures might mimic some features of LGS. 25 However, vitamin B6 response is seen as early as less than one week after begin of administration and further treatment with AEDs should not been postponed if a clear seizure reduction is not observed during this period.
Medical Therapy of LGS Based on Randomized Controlled Trials
Rufinamide is a new, orally active AED, which is effective in the treatment of partial seizures and drop attacks associated with LGS.36,37 It is well absorbed in the lower dose range, with approximately dose-proportional plasma concentrations up to 1,600 mg/day, but less than dose-proportional plasma concentrations at higher doses due to reduced oral bioavailability. 38 Steady state is reached within two days and its elimination half-life is 6–10 hours. A positive correlation has been identified between reduction in seizure frequency and steady-state plasma concentrations. The probability of adverse effects also appears to be concentration-related.
In a recent randomized placebo controlled trial it has been shown that rufinamide was effective for seizures associated with Lennox-Gastaut syndrome. 36 In this study, 139 patients were randomized with 138 patients receiving either rufinamide (n = 74) or placebo (n = 64) in addition to their other antiepileptic drugs. The median percentage reduction in total seizure frequency was significant greater in the rufinamide therapy group than in the placebo group (32.7% vs. 11.7%). When extrapolated to the different seizures types, there was a significant reduction in tonic-atonic (“drop attack”) seizures with rufinamide (42.5% median percentage reduction) when compared to placebo (1.4% increase). In addition, the rufinamide group had a greater improvement of seizure severity and a higher 50% responder rate compared with placebo for total seizures and tonic-atonic seizures. Common adverse events (reported by >or = 10% of patients receiving rufinamide) are somnolence and vomiting.
Topiramate is effective as an adjunctive therapy for refractory partial-onset seizures in adults. Subsequent studies indicate that topiramate is also effective as monotherapy in adults and as adjunctive therapy for partial-onset seizures in children, tonic-clonic seizures of non-focal origin in children and adults, and drop attacks in Lennox-Gastaut syndrome. Adverse effects for adults and children included dizziness, fatigue, ataxia, confusion, somnolence, nephrolithiasis, paresthesia, and weight loss. More adverse effects are observed at higher doses. Topiramate exhibits rapid absorption, long duration of action, and minimal interaction with other antiepileptic drugs. 39 Sachdeo and colleagues included ninety-eight patients > 1 year to <30 years of age with LGS in an 11-week, double-blind treatment phase with either topiramate or placebo. 40 The median percentage reduction of drop attacks from baseline in average monthly seizure rate was 14.8% for the topiramate group and -5.1% (an increase) for the placebo group. In addition, topiramate-treated patients demonstrated greater improvement in seizure severity than did placebo-treated patients. Thus, topiramate appears to be an effective adjunctive therapy in reducing the number of drop attacks and major motor seizures in patients with LGS.
Felbamate was the first of the new AEDs approved in the United States in 1993 with broad-spectrum efficacy against partial and generalized seizures of various types, and indicated for use as adjunctive and monotherapy. The identification of idiosyncratic aplastic anemia and hepatotoxicity, however, drastically curtailed its use. 41 The overall aplastic anemia risk is estimated between 27 and 209 per million in felbamate treated patients vs. 2 to 2.5 per million in the general population. The efficacy in LGS was evaluated in a randomized, placebo controlled trial. 42 73 patients ranging in age from 4 to 36 years who had LGS were included in the trial. The patients treated with felbamate had a 34% decrease in the frequency of atonic seizures, as compared with a 9% decrease in the patients who received placebo. The felbamate-treated patients had a 19% decrease in the total frequency of seizures, as compared with a 4% percent increase in the placebo group. In summary, the authors concluded that felbamate is beneficial in patients with the Lennox-Gastaut syndrome.
Lamotrigine is an antiepileptic drug of the phenyltriazine class. Lamotrigine's linear pharmacokinetics, low degree of protein binding, oral bioavailability, and activity in numerous animal models of seizures led to its development as a treatment for epilepsy. 43 Lamotrigine has a broad spectrum of activity and is effective in both partial and generalized seizures when used alone or with other antiepileptic drugs.44,45 It can also be given as adjunctive therapy for treatment-resistant epilepsy in childhood. 46 Besides evaluation in open-label studies, 47 Lamotrigine was evaluated in LGS patients in a 16-week, randomized, double-blind, placebo-controlled trial. 48 In this study, 169 patients were assigned to 16 weeks of lamotrigine (n = 79) or placebo (n = 90) in addition to their other antiepileptic drugs. The median frequency of all major seizures changed from base-line levels of 16.4 and 13.5 per week in the lamotrigine and placebo groups, respectively, to 9.9 and 14.2 per week after 16 weeks of treatment. Thirty-three percent of the patients in the lamotrigine group and 16 percent of those in the placebo group had a reduction of at least 50 percent in the frequency of seizures. There were no significant differences between groups in the incidence of adverse events, except for colds or viral illnesses, which were more common in the lamotrigine group. It was concluded the Lamotrigine was an effective and well-tolerated treatment for seizures associated with the Lennox-Gastaut syndrome.
Ketogenic Diet
The ketogenic diet (KD) consists of a high-fat, adequate-protein and low-carbohydrate diet and was developed in the 1920s based on observations of the benefits of fasting on seizure control. The mechanism of action of the ketogenic diet appears to rely on a fundamental change in the brain's metabolism from that of a glucose-based energy substrate to a ketone-based substrate. This change is, in some fashion, critical to the maintenance of seizure threshold. 49 The diet was successfully used during the 1920s and 1930s, but use declined after the introduction of a potent antiepileptic such as phenytoin. 50 Reports of KD efficacy and of the efficacy of the medium-chain triglyceride diet occasionally appeared during the 1970s and 1980s. In a KD pilot study with 17 patients with LGS, atonic or myoclonic seizures decreased in these patients by more than 50%. 50 These findings could be reproduced in a randomized cross over study though to a less significant extend.51,52 In conclusion, the KD is worth testing in refractory atonic and myoclonic seizure with LGS. However, some practical issue must be considered. The KD does only work when the diet is performed very strictly on the basis of the above mentioned high-fat, adequate-protein and low-carbohydrate intake. Thus, the diet is more difficult to establish in children who are capable of getting access to carbohydrate rich food as sweets or fruits. In addition to seizure reduction, the KD might be beneficial for behavioral aspects and the number of concomitant AEDs might be reduced. 25
Epilepsy Surgery
Vagal nerve stimulation has been shown to significantly reduce seizures in controlled studies to an extent similar to newly marketed anitepileptic drugs. Interestingly, further reduction of seizure frequency may occur with long term treatment even after 1 year. 53 In a number of studies implantation of vagal nerve stimulators resulted in a reduction of seizure frequency.54–56 As it appears usually in patients with vagal nerve stimulators as mentioned above, seizure reduction might be significant more after a one year period after implantation compared to a 6 months period after implantation. 57 Taken together, vagal nerve stimulators might be a promising approach in patients with LGS in whom medical treatment has failed to achieve sufficient seizure control. 58
Corpus callosotomy is performed in candidates with medically intractable LGS. Tonic and atonic seizures leading to injuries seem to respond better to callosotomy than other seizure types 59 which is also supported by the observation that it reduces drop attacks in LGS patients. 60 However, reduction of other seizures types associated with LGS in less common. Because some patients after complete callosotomy suffer from a disconnection syndrome, a staged approach has been developed. An anterior 2/3 callosotomy is performed first. If this fails to improve seizure control, complete disconnection can be performed in a second step usually without producing a disabling disconnection syndrome. 61 Corpus callosotomy was performed frequently in most major surgery of epilepsy centers in the early 1990s. However, the use of vagal nerve stimulation in patients with Lennox-Gastaut syndrome caused that many centers abandoned callosotomy or perform it only infrequently.
Resective epilepsy surgery might be the treatment of choice in localized cerebral lesions as tumors and vascular lesions though such lesion appear to be extremely rarely associated with LGS. 62
Treatment Algorithms for Patients with LGS
Some efforts have been made to establish treatment algorithms for patients with LGS.9,63 According to these approaches and to what was summarized the previous paragraphs, some practical recommendations for the clinical use can be made. Valproic acid remains the drug of choice for the initial treatment of LGS by most authorities. When seizure reduction is not sufficient, benzodiazepines may be added. If Vitamin B6 is considered, it should be tried in sufficient doses as a first line drug for not more than one week and should tapered quickly if no effect is observed. Typical second line drugs include AEDs who showed efficacy in randomized controlled trials as rufinamide, lamotrigine and topiramate. Although recent data might favor rufinamide among theses AEDs at this stage of management, finals remarks will depend on further experience and studies. 36 The use of felbamate is limited because of its risk of severe aplastic anemia. Vagal nerve stimulation or ketogenic diet can be considered when second line AEDs fail to achieve acceptable seizure reduction. Other AEDs and therapeutic procedures as corpus callosotomy can be tried in patients in whom treatment aims were not achieved with first or second line approaches.
Treatment of Status Epilepticus
In cases of generalized tonic-clonic status epilepticus, standard status epilepticus treatment guidelines involving benzodiazepines and phenytoin are mandatory. In contrast, in status like increase of other seizures types of LGS, the use of benzodiazepines or corticosteroids seems useful. However, benzodiazepines have to be used cautiously as they may precipitate tonic status epilepticus. 64
Supportive Care
The intractability of the seizures and the accompanied dysfunctions of cognition and behavior underline the importance of a multidisciplinary approach involving epileptologists, epilepsy nurses, social workers, psychologists and primary care physicians. An individually tailored treatment plan is mandatory in the majority of patients. In addition, the needs of parents and siblings have to be approached to avoid secondary problems in the family.
Prognosis
The prognosis for LGS is poor for both cognitive development and seizure outcome. The majority of patients with LGS will require lifelong assistance in their daily activities with some becoming wheelchair dependent.7,65 Less than 10% of patients with LGS will become seizure free in childhood or adolescence and may show no or only minor disability. Seizures in patients with LGS may be relatively well controlled on antiepileptic drug (AED) medication initially, but resistance to drugs usually develops over time. Ten years after diagnosis, seizures persist on a daily or weekly basis in approximately two-thirds of patients.
Poor prognostic indicators include the following:
Preceding West syndrome 66
High seizure frequency and repeated episodes of status epilepticus19,67
Persistence of diffuse slowing of the background and generalized SSWC 8
In conclusion, the availability of randomized controlled trials has offered new perspectives for some patients with LGS when first line drugs do not lead to convenient control of disabling seizures. On the site of epilepsy surgery procedures, vagal nerve stimulation can be beneficial in patients in whom medical treatment failed. Nevertheless, LGS remains a severe epileptic encephalopathy associated with life long disability in the majority of patients who need a multidisciplinary treatment approach to achieve the best results for the patients and their families.
Disclosures
The authors report no conflict of interest.