
Abstract
The voltage-gated sodium channel neuronal type 2 alpha subunit (Navα1.2) encoded by the SCN2A gene causes early infantile epileptic encephalopathy (EIEE) inherited in an autosomal dominant manner. Clinically, it has variable presentations, ranging from benign familial infantile seizures (BFIS) to severe EIEE. Diagnosis is achieved through molecular DNA testing of the SCN2A gene. Herein, we report on a 30-month-old Saudi girl who presented on the fourth day of life with EIEE, normal brain magnetic resonance imaging (MRI), normal electroencephalography (EEG), and well-controlled seizures. Genetic investigation revealed a novel homozygous missense mutation (c.5242A > G; p.Asn1748Asp) in the SCN2A gene (NM_001040142.1). This is the first reported autosomal recessive inheritance of a disease allele in the SCN2A and therefore expands the molecular and inheritance spectrum of the SCN2A gene defects.
Keywords
Introduction
The sodium channel neuronal type 2 alpha subunit (SCN2A) gene encodes the voltage-gated sodium channel Navα1.2 which is mainly responsible for the generation of action potentials in excitable cells. 1 Any mutation that causes loss of function (LOF) or changes the secondary structure of the SCN2A protein might result in the disruption of the sodium channel function. This might cause loss or decreased excitability of the neuronal cells, which contributes to autism spectrum disorder, or might cause hyperexcitability of neuronal cells, which is characterized as infantile seizures. 2 However, LOF, which does not result in hyperexcitable neurons, can still cause severe phenotypes with seizures. 3 Later in development, reduced excitability of unmyelinated cortical inhibitory neurons might lead to hyperexcitable cortical networks. Any pathogenesis due to SCN2A LOF might affect inhibitory/excitatory balance, which gives rise to seizures, neuropsychiatric disorders, and autism. 3
The most common clinical presentation of SCN2A defect is the benign familial infantile seizures type 3 (BFIS3; MIM 607745), which is an autosomal dominant neurological disorder, characterized by apnea, cyanosis, and cluster seizures that occur over one or several days. 4 The seizures are mostly focal, consisting of twitching of the limbs with the head turned, lips smacking, blinking of the eye, and staring episodes.5,6 Patients may also present secondary generalized seizures. 6 Usually, seizures cease at the end of the first year of life, yet some continue to have seizures through adulthood without any subsequent abnormality in neurological development.5,6 Electroencephalography (EEG) and magnetic resonance imaging (MRI) in these patients have insignificant results. Another phenotype associated with SCN2A pathogenesis include the early infantile epileptic encephalopathy type 11 (EIEE11), having autosomal dominant inheritance of the neurological disease, exhibiting a more severe neurological manifestation.7,8 Patients with EIEE present early onset of infantile refractory seizures, which eventually lead to a marked delay in intellectual and motor development.7,9 Initially, patients may present features such as neonatal hypotonia that proceeds to partial seizure along with generalized refractory tonic-clonic seizures, which might further progress to Ohtahara and West syndromes.10,11 Patients might exhibit dysphagia, dysarthria, excessive daytime sleepiness, disturbed visual contact, and in severe cases, paralysis.7,8,10 Brain MRI of these patients can show a variety of findings, with brain atrophy commonly reported.9,10,12 As with MRI, EEG findings vary among patients, mostly showing hypsarrhythmia and focal or multifocal sharp waves.7,8,12
To our knowledge, all the previously reported cases of SCN2A gene defects are inherited in dominant heterozygous state or de novo state, and there is no previously reported case having an autosomal recessive inheritance having homozygous variant in the SCN2A gene. Herein, we report the first patient with a biallelic missense mutation in the SCN2A gene with an autosomal recessive inheritance.
Case Report
The patient is a 30-month-old Saudi girl born full term via elective cesarean section to first cousin parents with an uneventful pregnancy. She is the fourth child of her parents with three older siblings. At birth, she had a length of 48 cm (10th-25th centile), weight of 3 kg (10th-25th centile), head circumference (HC) of 34 cm (10th-25th centile), and Apgar scores of 9 and 10 at 1 and 5 minutes, respectively. After discharge at 4 days of age, she presented with neonatal seizures described as mouth and eye deviation and jerking movement of the upper and lower limbs for one minute. At 1 month of age, the proband was alert, awake, afebrile, and she was prescribed phenobarbital and phenytoin after initiating appropriate investigations, including septic work up, brain computed tomography (CT), lumber puncture, EEG, and MRI of the brain. A week later, patient seizures were determined to be controlled on phenytoin and phenobarbital, and she was discharged as a follow-up patient with the pediatric neurology department. Later, she presented with multiple emergency department (ED) visits complaining mainly of seizures ranging from blinking and spasmodic hiccups to classical tonic-clonic movement, upon which she was electively admitted for further investigation, including long-term EEG monitoring and genetic consultation. Abnormal EEG results led to the addition of levetiracetam to the previous regimen.
Follow-up examination
Other than the symptoms mentioned above, the patient is thriving appropriately for her age and has no other medical problems. On examination at 30 months of age, her measurements were as follows: height 87 cm (10th-25th centile), weight 12 kg (10th-25th centile), and HC 48 cm (25th-50th centile). The patient generally looks well; is awake, alert, and active; and is not dysmorphic, with normal tone and power and normal gait and coordination. She is vitally stable and has unremarkable central nervous system (CNS) and other system examination results.
Multiple EEGs were performed, all with normal findings. Pediatric long-term monitoring (LTM) initially revealed abnormal sleep and awake readings, yet subsequent repeated LTM/EEGs were within normal limits. Brain MRI, abdominal ultrasound, cystourethrogram, renal ultrasound, auditory brainstem response, echocardiogram, and ophthalmology evaluations were normal. Complete blood count, serum drug and toxicity levels, renal profile, liver profile, and cerebrospinal fluid (CSF) assays for cell counts, protein, and glucose levels, stool and urine analyses were all normal.
Laboratory test
All of the following biochemical and genetic investigations were unremarkable: acylcarnitine profile, ammonia, lactic acid, creatine kinase, total homocysteine, urine amino acids, urine organic acids, coagulation profile, lipid profile, very long chain fatty acids, urine for creatine and guanidinoacetate, urine for purine and pyrimidines, chromosomal analysis, and comparative genomic hybridization (CGH) microarray.
Genetic and Molecular Analysis
Genomic DNA extraction
The present study was performed after the approval of institutional review board (IRB), followed by Helsinki protocols, and written informed consent was obtained from the patient’s parents to publish this case report. Genomic DNA was extracted using commercially available kit and quantified using standard methods.
Gene panel
Genetic testing with an EIEE gene panel (Centogene, Germany) identified a homozygous missense mutation (c.5242A > G; p.Asn1748Asp) in SCN2A (NM_001040142.1) located on chromosome 2q24.3.
Sanger sequencing
Segregation analysis of the identified homozygous variant was undertaken for both the parents, three siblings using standard methods, and showed that the parents and two siblings were heterozygous carriers, while one healthy sister is wild type.
The parents were comprehensively counseled regarding the genetic results, its mode of inheritance, and the recurrence risk of 25% in each pregnancy. The patient’s seizures are well controlled with phenobarbital and levetiracetam, and she has normal development with no deterioration or complications.
Discussion
The present report describes the first documented homozygous SCN2A mutation causing EIEE in autosomal recessive manner, as all previously reported patients were reported having heterozygous SCN2A variant in either dominant or de novo fashion. The pathogenicity of the variant (c.5242A > G; p.Asn1748Asp) reported here is supported by segregation within the family. Furthermore, the variant is rare and not described in homozygous or heterozygous state in the Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), Exome Variant Server (EVS), or 1000 Genomes Project and in 2000 in-house ethnically matched exomes. In addition, computational analysis tools, including PolyPhen, SIFT, MutationTaster, and phyloP, predict this variant to be most likely damaging.
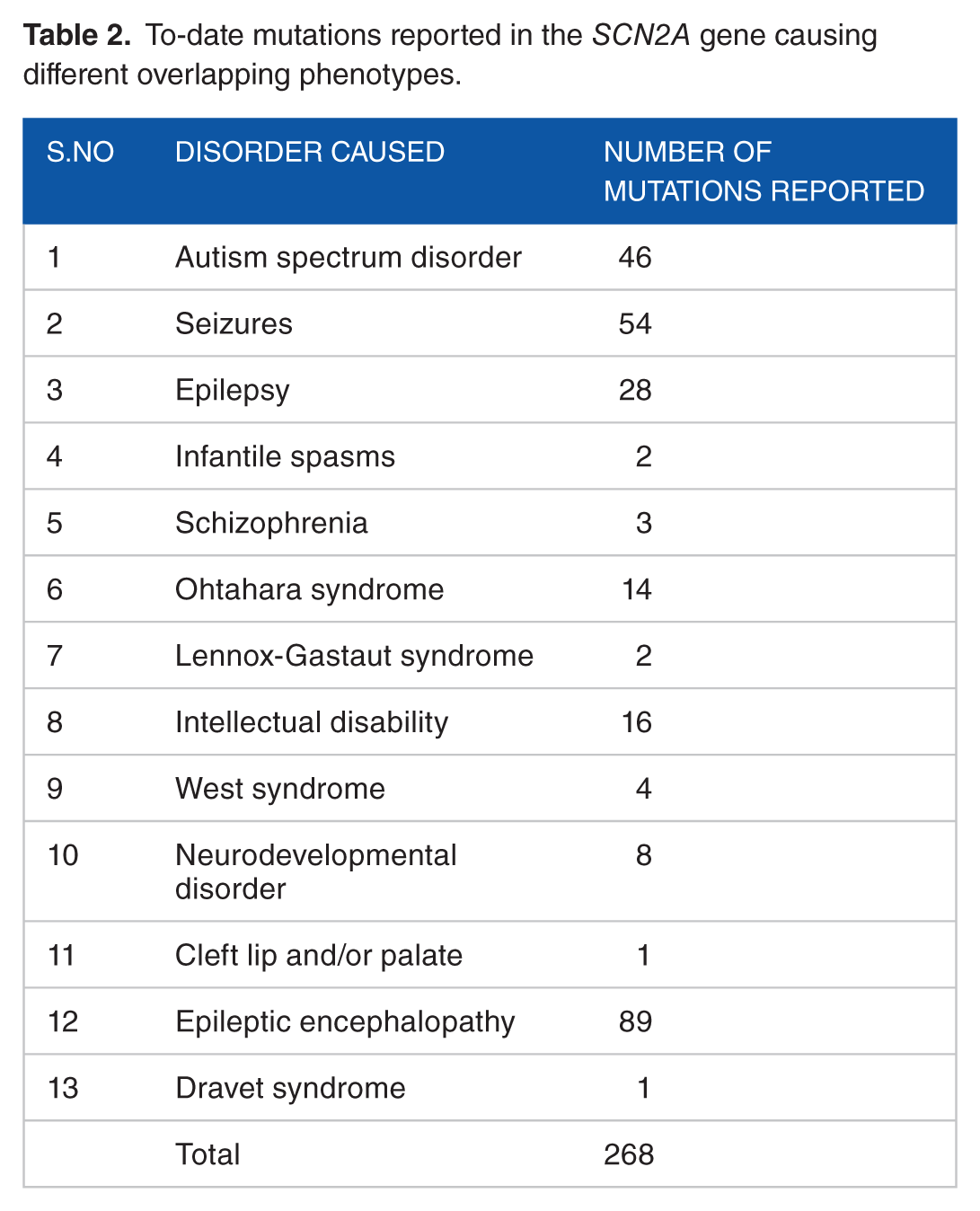
Pathogenic heterozygous and de novo mutations in the SCN2A gene have been reported with two causes: EIEE11 (MIM 613721) and BFIS3 (MIM 607745). Table 1 summarizes the patients reported to have EIEE due to either heterozygous or de novo mutations within the SCN2A gene.7,12–90 According to Human Gene Mutation Database (HGMD, 2018), 268 mutations have been reported in the SCN2A gene presenting diverse phenotypes (Table 2). We identified a total of 290 cases having heterozygous or de novo SCN2A gene mutations in the literature survey. Approximately 87.24% of cases reported several types of seizures, including BFIS, benign familial neonatal epilepsy, refractory neonatal epilepsy, infantile spasm, episodic ataxia, acute encephalopathy, recurrent encephalopathy, and EIEE, as observed in the present case. Other reported symptoms included autism (21.3%), severe pulmonary emphysema (4.48%), migrating focal seizures of infancy (6.55%), paroxysmal ataxia in toddler (4.48%), developmental delay (31.37%), Ohtahara syndrome (4.85%), West syndrome (1.37%), and Lennox-Gastaut syndrome (0.68%). The ages of symptom onset ranged from a few hours after birth up to 6.6 years. None of the previously reported cases were documented with parental consanguinity as described in our case. Regarding clinical features, 14.48% had normal features, 24.82% presented with hypotonia, 7.93% with ataxic gait, and 3.10% with spasticity. EEGs showed normal findings in 12.75%, including the current patient. Brain MRI was normal in our patient and in 19.31% of cases in literature. Moreover, 27.93% demonstrated cerebral atrophy, 13.44% showed hypoplastic corpus callosum, 18.27% suffered from cerebellar atrophy, and 3.10% had lesions in the right parietal/temporal/occipital lobes. The following features were observed in 5.86% of cases such as cortical/subcortical edema, diffuse abnormalities of white matter, lack of differentiation between cortex and subcortical layers with pachygyria and anomalies of the cortical gyration, and immature myelination at the bilateral periventricular areas. Up to 47.90% of cases were reported to have a family history of seizures; however, the current case had a negative family history of seizures. In total, 36 (12.41%) of cases reported so far were deceased, and the age at death ranged from 5 to 75 years old.

To-date mutations reported in the SCN2A gene causing different overlapping phenotypes.
Patient with de novo or dominant heterozygous SCN2A gene mutations display epileptic phenotypes. Thus, it is not surprising that patients with a homozygous SCN2A mutation also have an epileptic phenotype. Despite the aforementioned fact, the importance of the current case relies on the following: first, this alerts the clinician to consider SCN2A gene defects in any patient presenting with seizures regardless of their family history, suggesting an autosomal recessive or autosomal dominant pattern. Second, it is confirmed that an autosomal recessive pattern may lead to more severe presentation, which is early neonatal epileptic encephalopathy rather than ataxia and autism in heterozygous probands.
Mutations in the SCN2A gene have a strong association with the manifestation of numerous types of seizures, along with other clinical characteristics, including autism, pulmonary emphysema, paroxysmal ataxia in toddlers, and Ohtahara syndrome. However, mutations in the SCN1A, SCN1B, SCN8A, or SCN9A genes also appear to be related to the expression of seizures. Here, we identified a novel homozygous missense mutation in the SCN2A causing EIEE; yet heterozygous SCN2A mutations can cause severe epileptic phenotypes, especially when they are de novo. Therefore, clinicians should keep in mind the SCN2A gene heterogeneity. This is also true for mutations in the SCN1A gene, with de novo mutations causing the severe Dravet syndrome. Similarly, mutations in the SCN9A have been associated with pain disorders and suggested as a genetic modifier for the Dravet syndrome, thus considered as a susceptible gene.39-45 Moreover, SCN8A mutations are also inherited dominantly and associated with more severe manifestations of EIEE and BFIS.46,47 Homozygous SCN1B mutations have been shown to cause a clinical phenotype of Dravet syndrome, while heterozygous mutations in autosomal dominant cases have been associated with generalized epilepsy and febrile seizures.48,49 The limitation of the current study is being the only homozygous case described in the family and current literature making a firm inference of pathogenicity impossible; therefore, further homozygous reported cases with similar phenotype would confirm such conclusion.
Conclusion
We report the first case of a homozygous SCN2A gene mutation in a female toddler from Saudi Arabia having hallmark features of EIEE in autosomal recessive manner. In addition, we describe a novel mutation that increased the mutational spectrum of SCN2A-associated pathogenesis. This finding expands the molecular and inheritance spectrum of SCN2A gene defects.
Footnotes
Acknowledgements
The authors are grateful to the patient and her family for their genuine support.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Author Contributions
SAS wrote the first draft and edited subsequent versions. MAF and MU wrote the final draft, edited the subsequent version, and contributed to the clinical diagnosis and management of the patients. All authors have read and approved the final draft of the manuscript.