
Abstract
Developmental and epileptic encephalopathies (DEEs), such as SYNGAP1-related DEE, are marked by severe developmental delays and pharmaco-resistant seizures due to specific genetic variants. This case report focuses on a 9-year-old male with a de novo SYNGAP1 variant (c.1267del, p.Tyr423Metfs*17), illustrating the diagnostic and treatment challenges. Initially experiencing developmental delays and later, misdiagnosed tics, he was diagnosed with epilepsy with eyelid myoclonia at seven. His case includes key SYNGAP1 encephalopathy symptoms: intellectual disability, behavioral issues, and generalized epilepsy resistant to antiseizure medication. The identification of a specific variant adds to our knowledge, suggesting the necessity of considering SYNGAP1-related DEE for unexplained neurodevelopmental delays and seizures. This case underlines the need for a personalized treatment approach focusing on quality of life and symptom management, advancing our understanding and treatment practices for genetic developmental and epileptic encephalopathy.
Introduction
Developmental and epileptic encephalopathies (DEEs) refer to a group of severe epileptic disorders in which both an underlying etiology and frequent epileptic activity (such as seizures and abnormal electroencephalographic patterns) contribute to cognitive, neurological, and psychiatric deficits. 1 Among these, SYNGAP1-related DEE stands out due to its genetic etiology and complex clinical presentation. SYNGAP1, a gene located on chromosome 6p21.32, encodes a synaptic Ras GTPase Activating Protein 1 (SynGAP) critical for synaptic function and plasticity. 2 The prevalence of SYNGAP1 pathogenic variants in two studies was from 0.75% to 1% in two series of 931 and 500 individuals with DEE and unrelated children with intellectual disability.3,4
SYNGAP1-related DEE is characterized by developmental delay or intellectual disability, generalized epilepsy, and autism spectrum disorder (ASD). 5 The phenotypic variability and genetic heterogeneity observed among patients underscore the importance of individual case studies. These reports contribute significantly to the growing body of evidence, elucidating the natural history, genotype-phenotype correlations, and treatment responses in this population.
This case report aims to enhance understanding of SYNGAP1-related DEE by detailing a patient's clinical progression, management, and genetic findings.
Case presentation
We present a case of a 9-year-old male, born to non-consanguineous parents (father aged 34 and mother aged 31), with no relevant family history of genetic conditions associated with the symptoms presented. He was delivered by cesarean section at full term (40 weeks of gestational age) after a normal pregnancy, during which fetal movements had been reported as normal. At birth, his weight, head circumference, and length were normal. His APGAR was 9/9 and he exhibited adequate suction with no respiratory effort. No significant perinatal events were reported. However, the mother noted that the thumbs were flexed and abducted.
The parents report that the child did not hold their head up at 3 months, was unable to sit up unassisted at 6 months, and appeared developmentally delayed compared to children of the same age. They also reported slight irritability and limited eye contact with 1 year olds. Over the following months, psychomotor delay became more pronounced. At 2 years old, his parents sought medical attention due to the absence of language and difficulty walking. Additionally, they observed restricted interests, further reduced eye contact, low frustration tolerance, and frequent unusual eye movements (blinking and upward eye deviation), which they referred to as “tics”.
After consulting several doctors, he was diagnosed with ASD and continued without improvement for several years until he was brought to our clinic at the age of seven. The patient had been receiving speech, physical, and occupational therapy twice a week since the age of two. Examination revealed a normal weight, height, and head circumference for his age and sex, significant language development delay, absence of verbal communication, and difficulties in social interaction. Repetitive and stereotyped behaviors (hand flapping and body rocking), as well as restricted interests, were observed. Additionally, the patient showed avoidance of eye contact. Some self-injurious behaviors and aggression toward others were registered. Regarding the motor examination, no abnormalities in muscle tone, reflexes, or strength were observed; however, the patient presented with mild ataxia and walked on tiptoes.
During the consultation, ocular episodes were also observed, and it was noted that he occasionally lost generalized muscle tone, sometimes leading to falls. No provoking factors, such as mastication or chewing, were observed or reported by his parents. Suspecting eyelid myoclonia and drop-attacks, a 1-h sleep-only electroencephalogram (EEG) was performed, which revealed frequent generalized epileptiform discharges with spike-wave and polyspike-wave morphology. However, no clinical events were recorded. A previous 1.5T MRI had reported signs of frontal cortico-subcortical atrophy and a left temporal arachnoid cyst. Auditory evoked potentials were also reported as normal.
Given the suspicion of a genetic developmental and epileptic encephalopaty (DEE) and a previous normal 450 bands karyotype, he was referred to the Genetics service, where some dysmorphic features were identified in the physical exam (Table 1). A genetic panel for epilepsy and neurodevelopmental conditions was performed concerning 367 genes using the next-generation sequencing technique. Genomic DNA was obtained from a blood sample and was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology. Unless otherwise indicated, all targeted regions were sequenced with ⩾50× depth or supplemented with additional analysis. Reads were aligned to a reference sequence (GRCh37), and sequence changes were identified and interpreted in the context of a single clinically relevant transcript. A pathogenic variant, c.1267del (p.Tyr423Metfs*17), was identified in the SYNGAP1 gene. Both parents were also studied as a part of the genetic counseling without variants, proving the de novo origin of the variant. Treatment with valproic acid was initiated, and he was admitted to the epilepsy monitoring unit for electroclinical characterization of the ocular episodes. During the monitoring period, we recorded more than 50 episodes per day of blinking with upward eye deviation, lasting 3 s each, with an electroencephalographic correlate of generalized 3 Hz polyspike-wave discharges anteriorly predominant, consistent with generalized motor epileptic seizures of eyelid myoclonia (Supplemental Videos 1 and 2). We failed to record generalized episodes leading to falls.
Phenotypical signs present in our patient.
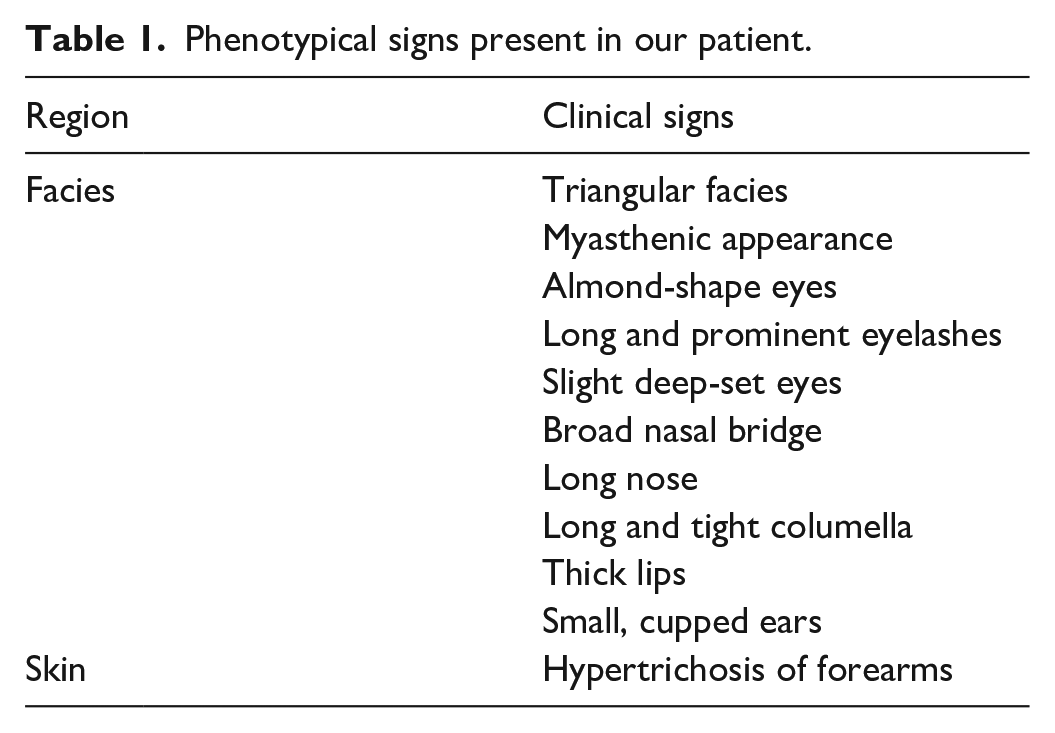
Eyelid myoclonia occurred exclusively during wakefulness and did not intensify during intermittent photic stimulation. Interictal epileptiform discharges were slightly activated during sleep, but not to the extent necessary to diagnose spike-and-wave activation during sleep. Despite increasing the dose of valproic acid, the frequency of eyelid myoclonia did not decrease, and side effects such as drowsiness and weight gain appeared. Therefore, the dose was reduced, and clobazam was added as adjunct therapy. Drop-attacks were controlled but eyelid myoclonia persisted according to parents’ reports. There was no formal neuropsychological evaluation, but parents reported a clinical improvement. For example, the patient started to follow commands, smile more, and look happier, he fell asleep earlier and without interruptions during the night, he no longer threw so many tantrums and self-injurious behaviors disappeared. Therefore, a joint decision was made with the parents not to increase the pharmacological treatment further. Currently, he continues to experience multiple daily eyelid myoclonia but without clinical deterioration.
Discussion
SynGAP is a brain-specific synaptic Ras GTPase activating protein predominantly localized to dendritic spines in neocortical pyramidal neurons. It plays a crucial role in suppressing signaling pathways linked to NMDA receptor (NMDAR)-mediated synaptic plasticity and is highly expressed at glutamatergic synapses. 2 The variant in exon 8, c.1267del (p.Tyr423Metfs*17), is heterozygous and this sequence change introduces a premature translational stop signal in the SYNGAP1 gene. This variant is predicted to cause a loss of function and those changes are known to be pathogenic because they are expected to result in an absent or disrupted protein product, thereby causing haploinsufficiency affecting the RASGAP domain of the protein. 6
According to the American College of Medical Genetics and Genomics guidelines, this variant is classified as PVS1, indicating very strong evidence of a pathogenic effect due to likely loss of function. Population data reveal that the variant's frequency is below the recommended threshold for this gene (0.05%), meeting the PM2 criterion. Moderate evidence supports its de novo origin, classified as PM6. Data from a reputable source provide additional support (PP5), and in silico predictions suggest a deleterious impact on protein function (PP3). Functional evidence places the variant in a mutational hotspot and/or critical functional domain (PM1), and it occurs in a gene with a low rate of benign missense mutations, where missense mutations are a common mechanism of disease (PP2). Phenotypically, the variant aligns with known clinical presentations, supporting a PP4 classification. Case-control studies provide strong evidence for association with the disease, meeting the PS4 criterion, and segregation data further support the variant’s pathogenicity (PP1), though in some cases, benignity is considered (BS4).
This variant has not been reported previously in population databases as GnomAD (no frequency) and it has not been reported in the literature in individuals affected with SYNGAP1-related conditions. ClinVar contains a report of a patient with the same mutation (Accession: SCV003325063.2); however, considering the date of the report and the laboratory providing the technique, it must be the same case.
Our patient has a variant in exon 8 and presents with pharmacoresistant epilepsy and severe language impairment. Some authors have reported that patients with variants in exons 1–4 were more likely to have the ability to speak in phrases vs those with variants in exons 5–19, and epilepsy occurred less frequently in patients with variants in the SH3 binding motif. 7 Seizures in patients with variants in exons 4–5 were more pharmacoresponsive than in patients with variants in exons 8–15. 5
The diagnosis of epilepsy in this patient was not established until the age of seven, because eyelid myoclonia, although present since the age of two, were misinterpreted as tics. Eyelid myoclonia is one the most common seizure types reported in patients with SYNGAP1-related DEE, and its confusion with tics is a well-recognized issue in pediatric patients. 8 Previous studies have indicated a predominance of absence seizures and drop-attacks.5,9 We failed to record drop-attacks during epilepsy monitoring unit (EMU) admission, possibly due to the initiation of antiseizure medication; however, eyelid myoclonia persisted.
Some authors have reported novel seizure types in these patients. For example, Vlaskamp et al. reported five patients with eyelid myoclonia–myoclonic–atonic seizures and eight patients with eyelid myoclonia–atonic seizures in a series of 36 patients with SYNGAP1-DEE and eyelid myoclonia. 9 We cannot confirm whether our patient presents with this same epilepsy phenotype due to the absence of drop-attacks during EMU admission, and the fall observed during consultation was too brief. However, the parents reported occasionally observing an association between ocular movements and falls.
Previous reports have shown that 40%–100% of SYNGAP1 patients develop pharmacoresistant epilepsy.5,7,9–11 Our patient continued to exhibit multiple daily episodes of eyelid myoclonia despite increased dosage and the addition of new adjunct therapies. Despite this, he demonstrated behavioral and cognitive improvements. During the long-term video-EEG, we confirmed normal sleep architecture despite the presence of interictal epileptiform discharges, a finding associated with favorable developmental outcomes. 12 A recent report from an international expert panel showed a strong consensus that the goals of the treatment would allow for ongoing eyelid myoclonia as long as all other seizure types were controlled. 13 Therefore, we decided to maintain close monitoring, continue speech, physical, and occupational therapy, and opted not to escalate antiseizure medication.
In our patient’s EEG findings, both ictal and interictal abnormalities presented as 3 Hz spike-and-wave and polyspike-and-wave epileptiform discharges, which were bilateral and synchronous. The posterior dominant rhythm was also slower (6 Hz) than expected for his age. These findings are in line with previous EEG studies in patients with SYNGAP1-related DEEs.11,14,15 Interestingly, although photosensitivity has been reported in many patients,5,7,11,14,15 we failed to confirm the presence of a photoparoxysmal response during intermittent photic stimulation (IPS). Some authors have demonstrated that IPS exhibits its maximal sensitivity when conducted according to the ILAE recommendations, which include performing the procedure at every frequency in three different states: eyes open, eye closure, and eyes closed. 16 Due to our patient's uncooperative condition, we were unable to adhere to this protocol. Consequently, we cannot completely exclude the possibility of photosensitivity.
Conclusion
Our findings underscore the critical need for awareness among clinicians regarding the diverse manifestations of SYNGAP1 variants, particularly the potential for misdiagnosis of epileptic seizures as tics, as observed with eyelid myoclonia in this patient. This case also exemplifies the challenge of managing pharmacoresistant epilepsy, common in SYNGAP1-related conditions, despite the use of antiseizure medications. The persistence of eyelid myoclonia despite treatment, combined with the patient's cognitive and behavioral improvements, led to a decision against aggressive treatment of seizures, focusing instead on quality of life and developmental outcomes. This report also adds to the genetic heterogeneity known to be associated with SYNGAP1 disorders and highlights the importance of genetic testing in atypical developmental and epileptic presentations.
Footnotes
Acknowledgements
We would like to extend our heartfelt gratitude to the patient’s parents for granting us permission to publish the case of their son. Their generous consent will not only contribute to the medical community’s understanding but also help improve the care of similar patients worldwide. Their cooperation and willingness to share their son’s story are invaluable, and we deeply appreciate their support in advancing medical knowledge.
Author contributions
N.E.L.O. conceptualization, data curation, writing–original draft preparation; F.V.V.V. data curation, writing–original draft preparation; J.C.B.L. and J.A.C.O. resources, writing–reviewing and editing; F.A.M. and T.P.S.M. writing–reviewing and editing. A.G.A. supervision, writing–reviewing and editing, visualization and project administration. All authors discussed the results and commented on the manuscript.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics approval
Our institution does not require ethical approval for reporting anonymized individual cases or case series.
Informed consent
Complete written and verbal informed consent was obtained from the patient’s relatives for conducting and publication of the study with accompanying videos and images.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.