
Abstract
Developmental and epileptic encephalopathies (DEEs) are rare neurodevelopmental disorders characterised by early-onset and often intractable seizures and developmental delay/regression, and include Dravet syndrome and Lennox–Gastaut syndrome (LGS). Rufinamide, fenfluramine, stiripentol, cannabidiol and ganaxolone are antiseizure medications (ASMs) with diverse mechanisms of action that have been approved for treating specific DEEs. Rufinamide is thought to suppress neuronal hyperexcitability by preventing the functional recycling of voltage-gated sodium channels from the inactivated to resting state. It is licensed for adjunctive treatment of seizures associated with LGS. Fenfluramine increases extracellular serotonin levels and may reduce seizures via activation of specific serotonin receptors and positive modulation of the sigma-1 receptor. Fenfluramine is licensed for adjunctive treatment of seizures associated with Dravet syndrome and LGS. Stiripentol is a positive allosteric modulator of type-A gamma-aminobutyric acid (GABAA) receptors. As a broad-spectrum inhibitor of cytochrome P450 enzymes, its antiseizure effects may additionally arise through pharmacokinetic interactions with co-administered ASMs. Stiripentol is licensed for treating seizures associated with Dravet syndrome in patients taking clobazam and/or valproate. The mechanism(s) of action of cannabidiol remains largely unclear although multiple targets have been proposed, including transient receptor potential vanilloid 1, G protein-coupled receptor 55 and equilibrative nucleoside transporter 1. Cannabidiol is licensed as adjunctive treatment in conjunction with clobazam for seizures associated with Dravet syndrome and LGS, and as adjunctive treatment of seizures associated with tuberous sclerosis complex. Like stiripentol, ganaxolone is a positive allosteric modulator at GABAA receptors. It has recently been licensed in the USA for the treatment of seizures associated with cyclin-dependent kinase-like 5 deficiency disorder. Greater understanding of the causes of DEEs has driven research into the potential use of other novel and repurposed agents. Putative ASMs currently in clinical development for use in DEEs include soticlestat, carisbamate, verapamil, radiprodil, clemizole and lorcaserin.
Keywords
Introduction
The International League Against Epilepsy (ILAE) defines ‘epileptic encephalopathy’ as a condition where ‘the epileptic activity itself may contribute to severe cognitive and behavioural impairments above and beyond what might be expected from the underlying pathology alone (e.g., cortical malformation), and that these can worsen over time’. 1 Although such impairments may have adverse effects on an individual’s development (e.g., by slowing developmental skills), the ILAE subsequently conceived the term ‘developmental and epileptic encephalopathy’ (DEE) to categorise conditions where the underlying pathology – often a deleterious genetic mutation – has developmental sequelae independent of those arising from the epileptic encephalopathy.2,3
DEEs encompass a heterogeneous group of rare neurodevelopmental disorders, characterised by early-onset and often intractable seizures, typically starting in infancy or childhood. They also present with developmental delay and/or regression and electroencephalographic abnormalities and, in some cases, are associated with early death.4,5 These disorders include West syndrome, Dravet syndrome, Lennox–Gastaut syndrome (LGS), Ohtahara syndrome, early myoclonic encephalopathy, epilepsy of infancy with migrating focal seizures, myoclonic-atonic epilepsy, focal lesional epilepsies (e.g., tuberous sclerosis complex, hemimegalencephaly), Rasmussen syndrome and Landau–Kleffner syndrome, with West syndrome accounting for over half of cases (Figure 1).3,6,7 Information on the incidence of DEEs is limited, but an epidemiological study conducted in the Wellington region of New Zealand among children aged <16 years presenting with epilepsy estimated the cumulative incidence of DEEs to be 169/100,000 children. 8 Although many DEEs are now thought to have a genetic aetiology, other causes are also implicated, including DEEs with structural (e.g., tuberous sclerosis complex), metabolic (e.g., glucose transporter-1 deficiency) and immune/inflammatory (e.g., Rasmussen syndrome) aetiologies.6,9–13
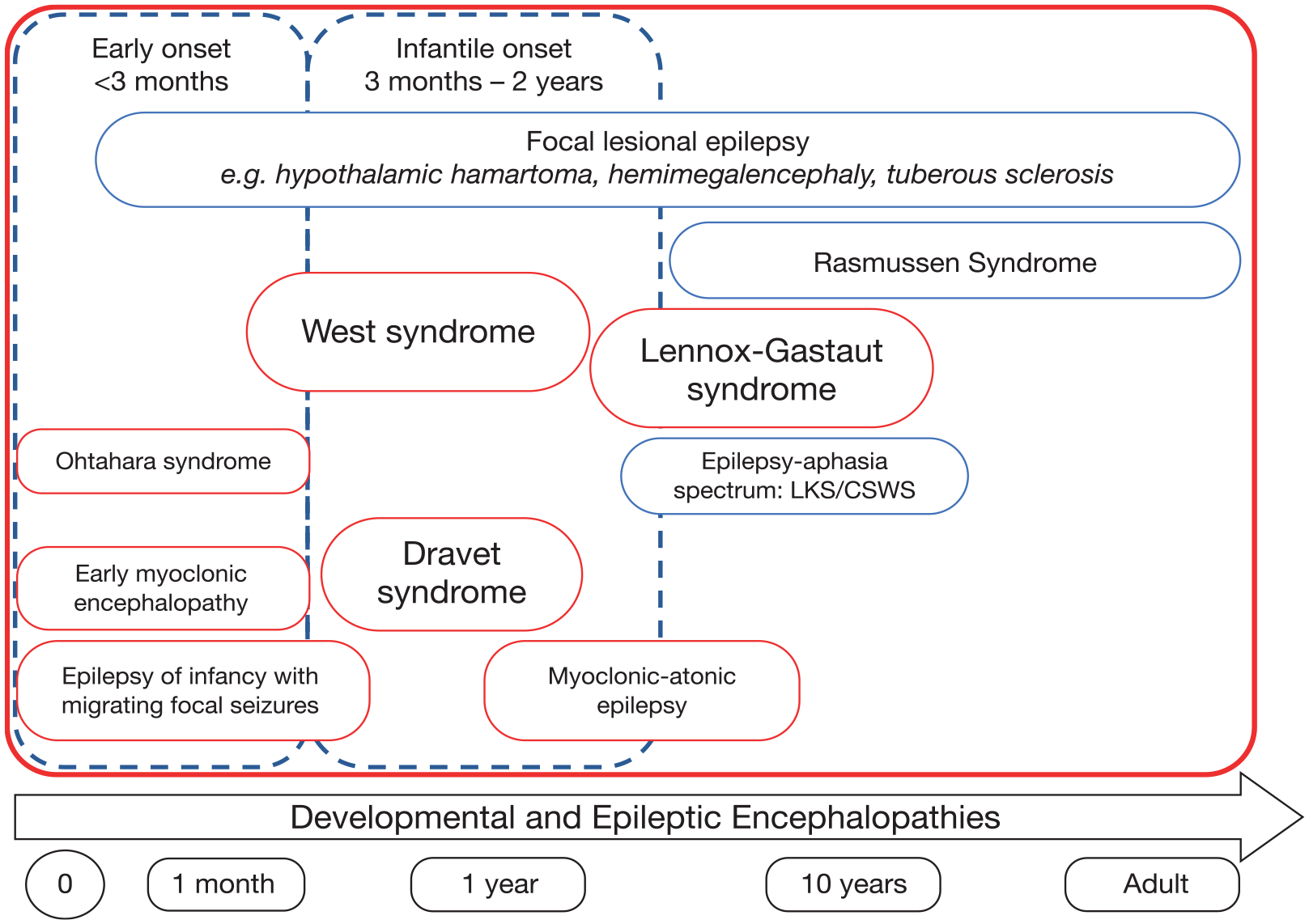
Schematic of developmental and epileptic encephalopathies (DEEs) with onset at different ages.
DEEs usually have onset during infancy or early childhood (Figure 1), although any age group may be affected, including adults. 3 Phenotypic presentation in different DEEs varies not only according to the underlying cause of the condition, but also during different phases of development and brain maturation, with the type and severity of the epilepsy potentially changing across the disease course. 9 DEEs are particularly challenging to treat and are often unresponsive to conventional antiseizure medications (ASMs), which may be explained, at least in part, by an intrinsically high frequency and severity of seizures.14–16 Even when seizures are controlled, patients are often left with severe neurological disability that has a substantial impact on their functioning and quality of life.14,15 Moreover, in addition to seizures, patients often experience a range of comorbidities, including intellectual disability, movement disorders, respiratory, gastrointestinal and orthopaedic issues and sleep problems, and have a significantly increased risk of Sudden Unexpected Death in Epilepsy (SUDEP).14,15 Early recognition and intervention are required in order to optimise outcomes for patients with DEEs. 9
Current therapeutic options for the treatment of seizures in DEEs
A broad range of conventional ASMs are currently deployed in the treatment of DEEs, due to the variety of seizure types involved and their inherent intractability.1,17–20 Improved understanding of the genetic aetiology of many DEEs, in terms of both the identification of the genes involved and the downstream sequelae of the respective genetic mutations, has the potential to facilitate the development of precision drugs to target the pathogenic mechanisms underlying a particular condition, not only reducing seizure occurrence but also targeting non-seizure symptoms.13,15,21,22 Moreover, increasing awareness of the role of neuroinflammation in DEEs raises the possibility of developing other types of targeted therapies to prevent seizure recurrence and developmental regression. 6 Other therapeutic approaches to the treatment of seizures in DEEs include vagus nerve stimulation, 23 surgery24–27 and use of the ketogenic diet.18,27–30 These alternative treatment options have been reviewed elsewhere and are out-with the scope of the current article. This review article will instead focus on the mechanisms of action, pharmacokinetics and clinical trials findings of five ASMs that have been specifically approved for use in DEEs, and on other emerging ASMs in this setting. The aim is to provide a comprehensive update on these drugs that draws specific attention to the diversity in their pharmacology.
Use of ASMs in the treatment of DEEs
Rufinamide, fenfluramine, stiripentol, cannabidiol and ganaxolone are ASMs with diverse mechanisms of action that have been approved for treating specific DEEs (Table 131–49).
Summary of pharmacology of ASMs used in the treatment of DEEs.

Absolute bioavailability is not known because an intravenous formulation is not available for testing.
Degrades rapidly in an acidic environment.
Cmax and AUC increase by three- and twofold, respectively, when administered with a high-fat meal, compared with fasted conditions.
After twice daily dosing for 7 days in healthy volunteers.
In healthy volunteers.
Central Vd/F in two-compartment pharmacokinetic analysis in a mouse model of treatment-resistant status epilepticus.
Co-administration with strong inducers of CYP1A2 or CYP2B6 may decrease fenfluramine plasma concentrations.
Based on in vitro studies.
When administered with clobazam, stiripentol increases plasma concentrations of clobazam (a substrate of CYP3A4) and the active metabolite of clobazam, norclobazam (a substrate of CYP2C19).
ASM, antiseizure medication; AUC, area under the time–plasma concentration curve; Cmax, maximum plasma concentration; CDKL5, cyclin-dependent kinase-like 5; CYP, cytochrome P450; DEE, developmental and epileptic encephalopathy; GABA, gamma-aminobutyric acid; GABAA, type-A GABA; GPR-55, G protein-coupled receptor 55; LGS, Lennox–Gastaut syndrome; MATE1, multidrug and toxin extrusion transporter 1; MoA, mechanism of action; mRNA, messenger ribonucleic acid; tmax, time to maximum plasma concentration; t½, plasma half-life; TRPV1, transient receptor potential vanilloid 1; TSC, tuberous sclerosis complex; UGT, uridine 5′-diphospho-glucuronosyltransferase; Vd/F, volume of distribution.
Rufinamide
Rufinamide (1-[2,6-difluoro-phenyl] methyl-1 hydro-1,2,3-triazole-4 carboxamide) was developed in the 1990s as a triazole derivative, 50 and is structurally unrelated to other commonly used ASMs. 51 It was initially granted orphan drug status in 2004 for the adjunctive treatment of seizures associated with LGS in patients aged ⩾4 years 52 and is currently licensed in several countries across Europe and in the United States, where it is approved for the adjunctive treatment of seizures associated with LGS in patients aged ⩾1 year.31,32 Rufinamide is also approved for the adjunctive treatment of seizures associated with LGS in patients ⩾4 years in Japan. 53
Pharmacokinetics and pharmacology
The pharmacokinetic profile of rufinamide is summarised in Table 1. Rufinamide does not inhibit the activity of cytochrome P450 (CYP) enzymes and does not appear to have a clinically relevant effect on steady-state concentrations of carbamazepine, lamotrigine, phenobarbital, topiramate, phenytoin or valproate. 31 However, it has a modest to moderate inducing effect on CYP3A4 and reduces the plasma concentrations of triazolam and oral contraceptives (ethinyl oestradiol and norethindrone). 31 Valproate significantly decreases the clearance of rufinamide, and a lower maximum dose of rufinamide is therefore recommended for patients treated concomitantly with valproate. 31
Although the precise mechanism of action of rufinamide is unclear, it is believed to act primarily by modulating sodium-dependent action potentials, specifically by slowing the functional recycling of voltage-gated sodium channels from the inactivated to resting state following depolarisation and thereby suppressing neuronal hyperexcitability.33,54 Rufinamide has been shown to slow sodium channel recovery from inactivation after a prolonged pre-pulse in rat cortical neurons, and to limit sustained repetitive firing of sodium-dependent action potentials, with both effects occurring at clinically relevant concentrations. 55 In addition to effects on the recovery of sodium channels from inactivation, rufinamide has been shown to inhibit activation of the human voltage-gated sodium channel type I alpha subunit (Nav1.1; SCN1A) in Xenopus oocytes. 56 Other preclinical studies have shown that rufinamide has no effect on gamma-aminobutyric acid (GABA) receptors, no appreciable binding to glutamatergic, serotonergic, histaminergic, adrenergic or muscarinic cholinergic receptors, and no effect on adenosine transport. 57
Preclinical and clinical evidence
In rodent models of focal and generalised seizures, rufinamide exhibits a broad spectrum of anticonvulsant activity, including suppression of maximal electroshock (MES)-induced tonic seizures and pentylenetetrazol (PTZ)-induced clonic seizures, 33 which is somewhat unusual for a drug that is a supposedly selective inhibitor of voltage-gated sodium channels. In MES tests conducted in mice, the ED50 for orally administered rufinamide was 23.9 mg/kg, compared with 9.0, 20.1, 664.8 and >2000 mg/kg for phenytoin, phenobarbital, valproate and ethosuximide, respectively. 33 In mouse PTZ tests, the ED50 for oral rufinamide was 45.8 mg/kg, compared with >300, 12.6, 388.3 and 192.7 mg/kg for phenytoin, phenobarbital, valproate and ethosuximide, respectively. 33 In addition, the behavioural toxicity of rufinamide, as determined by the rotarod test, was equivalent or superior to these comparator ASMs. 33
Rufinamide was licensed on the basis of a 12-week, multicentre, randomised, double-blind, placebo-controlled, parallel-group trial, conducted in patients with LGS aged 4–30 years, 58 and a long-term, open-label extension study, in which patients who completed the initial randomised trial received rufinamide for a median period of 432 days. 59 The initial trial showed that rufinamide was significantly more effective than placebo in reducing the frequency of drop attacks (tonic-atonic seizures) and total seizures, and significantly more effective than placebo in improving seizure severity. 58 Rufinamide was generally well tolerated, with the most frequently reported adverse events (AEs) (reported by ⩾10% of patients, higher incidence in rufinamide versus placebo groups) being somnolence and vomiting. 58 In the open-label extension, reductions in seizure frequency were sustained throughout the study period, and tolerability observed in the initial trial was maintained over the long term. 59 A subsequent phase IV, non-interventional, multicentre registry study, conducted in patients with LGS aged ⩾4 years, revealed no unexpected safety issues with rufinamide after a median duration of follow-up of > 2 years; the most frequently reported AEs were somnolence (7.8% of patients) and decreased appetite (6.3% of patients). 60
Fenfluramine
Fenfluramine (3-trifluoromethyl-N-ethylamphetamine) was originally developed in the 1960s as an appetite suppressant for the treatment of obesity. 61 However, it was later withdrawn from the market due to reports of a possible link with valvular heart disease and primary pulmonary hypertension. 62 Subsequently, low-dose fenfluramine was found to have antiseizure effects in patients with Dravet syndrome, with 70% (7/10) of participants in one retrospective analysis demonstrating seizure freedom for at least 1 year at the time of last clinic visit and 30% (3/10) of the same cohort maintaining seizure freedom for 5 years during long-term prospective follow-up.63–65 Fenfluramine was granted orphan drug status for the treatment of Dravet syndrome in 2013 66 and for the treatment of LGS in 2017. 67 Fenfluramine is currently licensed as adjunctive treatment for seizures associated with Dravet syndrome (European Union, United Kingdom, United States and Japan) and LGS (European Union and United States) in patients aged ⩾2 years.35,36,68
Pharmacokinetics and pharmacology
The pharmacokinetic profile of fenfluramine is summarised in Table 1. In vitro studies have indicated that fenfluramine may inhibit CYP2D6 and induce CYP2B6 and CYP3A4, and that co-administration of fenfluramine with strong inducers of CYP1A2 or CYP2B6 may decrease its plasma concentration.35,69,70 They also suggest that norfenfluramine (the major pharmacologically active metabolite of fenfluramine) may inhibit multidrug and toxin extrusion transporter 1 (MATE1) at clinically relevant concentrations, 35 resulting in a theoretical increase in plasma concentrations of MATE1 substrates (e.g., cimetidine, metformin). Co-administration of single-dose fenfluramine alongside single doses of stiripentol, clobazam and valproic acid was without pharmacokinetic effect; however, the presence of stiripentol has been reported to increase fenfluramine exposure and to decrease that of norfenfluramine, with consequent recommendations for maximal fenfluramine dosing. 35 Pharmacodynamic interactions between fenfluramine and serotonergic agents (including selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, tricyclic antidepressants and triptans) increase the risk of aggravated central nervous system (CNS) depression and serotonin syndrome. 35
Fenfluramine targets the serotonin [5-hydroxytryptamine (5-HT)] system and sigma-1 receptors.37,71–75 Fenfluramine and its active metabolite, norfenfluramine, are 5-HT-releasing agents that disrupt vesicular storage of serotonin in presynaptic nerve terminals and also reduce the reuptake of serotonin from the synaptic cleft by inhibition of serotonin transporter proteins. 71 The consequent elevation in synaptic concentrations of serotonin is anticipated to result in the activation of multiple 5-HT receptor subtypes. In addition, norfenfluramine appears to possess direct agonist effects at selected serotonin receptors, including 5-HT2B, 5-HT2C and 5-HT1D, whereas fenfluramine itself is an agonist at 5-HT1D, 5-HT2A, 5-HT2B and 5-HT2C receptors and a positive modulator of the sigma-1 receptor.37,72–75 The extent to which these, and other mechanisms, contribute to the antiseizure effects of fenfluramine remains the subject of ongoing investigation.35,36
Preclinical and clinical evidence
Most cases of Dravet syndrome are caused by de novo mutations in the voltage-gated sodium channel type I alpha subunit gene (SCN1A).76,77 The phenotype of homozygous scn1Lab mutant zebrafish larvae resembles that of Dravet syndrome, and this model has provided a means of studying the antiseizure effects of fenfluramine in vitro. 73 Using this model, and a range of selective ligands for a variety of receptors, fenfluramine was shown to exert its antiseizure effects via agonist action at 5-HT1D and 5-HT2C receptors73,74 (Figure 2). These studies also indicated that fenfluramine might additionally act on the sigma-1 receptor, since its antiepileptiform activity in scn1Lab mutants appeared to be diminished in the presence of a selective sigma-1 receptor ligand. 73 Subsequent in vitro and in vivo experiments have confirmed a role of the sigma-1 receptor in the pharmacology of fenfluramine, suggesting that its mechanism of action as an ASM may involve both enhancement of serotonergic neurotransmission and positive allosteric modulation of the sigma-1 receptor37,78 (Figure 2). This is reinforced by the ability of fenfluramine to potentiate the low-dose effects of neuroactive steroids, which are endogenous sigma-1 receptor modulators that potently modify the excitatory/inhibitory balance in the brain. 75

Serotonergic mechanisms of action of fenfluramine: (a) serotonergic (5-HT) modulation and (b) positive modulation of sigma1R.
The scn1Lab mutant zebrafish model has also been used to investigate mechanisms of epileptogenesis in Dravet syndrome and has provided evidence that chronic treatment with fenfluramine, but not diazepam, can restore dendritic arborisation of GABAergic neurons and correct cell hyperproliferation defects (e.g., astrogliosis) associated with the development of epileptiform activity in this model. 79 Whether fenfluramine possesses similar antiepileptogenic potential in the clinical setting is yet to be determined.
Fenfluramine was licensed for the adjunctive treatment of seizures in patients with Dravet syndrome on the basis of two phase III, randomised, placebo-controlled trials,80,81 and an open-label extension study. 82 In the first trial, children and young adults with Dravet syndrome (mean age, 9.0 years) were randomised (1:1:1) to receive fenfluramine 0.2 mg/kg/day, fenfluramine 0.7 mg/kg/day or placebo, added to existing ASMs (excluding stiripentol), for a total 14 weeks (2 weeks titration, 12 weeks maintenance). 80 Mean reduction in monthly convulsive seizure frequency was significantly greater for fenfluramine 0.7 mg/kg/day, when compared with placebo, and the most commonly reported AEs (⩾10% of patients, higher incidence in fenfluramine versus placebo groups) were decreased appetite, diarrhoea, fatigue, lethargy, somnolence and decreased weight. 80 In the second study, children and young adults with Dravet syndrome (mean age, 9.1 years) were randomised (1:1) to receive fenfluramine 0.4 mg/kg/day or placebo, added to existing ASMs (including stiripentol), during a 3-week titration phase and 12-week maintenance phase. 81 Reduction in mean monthly convulsive seizure frequency was significantly greater with fenfluramine versus placebo, as was the proportion of patients achieving ⩾50% reduction in monthly convulsive seizure frequency (54% fenfluramine, 5% placebo), an outcome measure that was considered ‘clinically meaningful’. 81 The most frequently reported AEs in patients treated with fenfluramine versus placebo were decreased appetite (44% versus 11%), fatigue (26% versus 5%), diarrhoea (23% versus 7%) and pyrexia (26% versus 9%). 81 Neither trial reported any clinical or echocardiographic evidence of valvular heart disease or pulmonary arterial hypertension.80,81
The longer-term efficacy and safety/tolerability of fenfluramine in Dravet syndrome have subsequently been established in an open-label extension study of patients who completed one of the phase III trials, with maximal doses of fenfluramine adjusted for the presence of stiripentol. 82 Efficacy was sustained over a median treatment period of 256 days, with no difference in outcome when the cohort was stratified by age. 82 Treatment-emergent AEs again included pyrexia (21.6% of participants) and decreased appetite (15.9% of participants) and cardiac monitoring was once more unremarkable. 82 In a subsequent assessment of cardiac safety in the open-label extension study, conducted after a median treatment duration of 23.9 months, there were no cases of valvular heart disease or pulmonary arterial hypertension. 83 A post-hoc pooled analysis of clinical trial data has demonstrated that the rates of all-cause and SUDEP mortality in patients with Dravet syndrome treated with fenfluramine were substantially lower than those reported historically for patients with Dravet syndrome treated with standard of care. 84 The longer-term efficacy and safety/tolerability of fenfluramine in Dravet syndrome have also been confirmed in real-world compassionate use and early access programmes, which have additionally demonstrated that up to 50% of patients were able to reduce or discontinue treatment with other ASMs following the introduction of fenfluramine.85,86
Fenfluramine was licensed for the adjunctive treatment of seizures in patients with LGS on the basis of a phase III, randomised, placebo-controlled trial, in which patients with LGS aged 2–35 years were randomised (1:1:1) to receive fenfluramine 0.2 mg/kg/day, fenfluramine 0.7 mg/kg/day or placebo, added to existing ASMs (excluding cannabidiol), for a total 14 weeks (2 weeks titration, 12 weeks maintenance). 87 Median reduction in monthly drop seizure frequency was significantly greater for fenfluramine 0.7 mg/kg/day versus placebo (particularly for generalised tonic-clonic seizures), and the most commonly reported AEs (⩾10% of patients, higher incidence in fenfluramine versus placebo groups) were decreased appetite, somnolence, fatigue, diarrhoea and vomiting. 87 In an interim analysis of the open-label extension to this trial (median treatment duration, 364 days), patients continued to experience sustained reductions in drop seizure frequency and fenfluramine was generally well tolerated. 88 There were no reported cases of valvular heart disease or pulmonary arterial hypertension in the initial trial or open-label extension study.87,88
Stiripentol
Stiripentol (4,4-dimethyl-1-[3,4-[methylenedioxy]-phenyl]-1-penten-3-ol) is an aromatic allylic alcohol that is structurally unrelated to other ASMs. 89 It was the first ASM to be granted orphan drug status by the European Medicines Agency in 2000 and was subsequently registered in Europe in 2007 as an orphan drug for the treatment of Dravet syndrome, as adjunctive therapy with valproate and clobazam. 90 Stiripentol is currently licensed in Europe for use as adjunctive therapy for refractory generalised tonic-clonic seizures in patients with Dravet syndrome whose seizures are not adequately controlled by existing treatment with a combination of clobazam and valproate. 40 In the United States, stiripentol is licensed for the treatment of seizures associated with Dravet syndrome in patients taking clobazam aged ⩾6 months and weighing ⩾7 kg. 41
Pharmacokinetics and pharmacology
The pharmacokinetic profile of stiripentol is summarised in Table 1. Stiripentol is a broad-spectrum inhibitor of several prominent hepatic drug metabolising enzymes, including CYP2C19, CYP3A4 and CYP2D6, and can potentially cause marked increases in the plasma concentrations of concomitant therapeutic agents metabolised by these enzymes. 40 Its own metabolism is catalysed by CYP1A2, CYP2C19 and CYP3A4, and caution is therefore also advised when combining stiripentol with drugs that inhibit or induce these enzymes. 40 Due to its effects on CYP enzymes, stiripentol may increase plasma levels of benzodiazepines (e.g., diazepam, midazolam, clobazam) and lead to excessive sedation and may also enhance the central depressant effect of chlorpromazine. 40 Similarly, stiripentol may enhance the effects of other ASMs, including phenobarbital, primidone, phenytoin, carbamazepine, valproate and ethosuximide, potentially necessitating clinical monitoring of plasma levels and dose adjustments. 40 Such dose adjustments do not appear to be required when stiripentol is used with topiramate or levetiracetam. 40 Stiripentol has, on occasion, been described as a ‘co-therapy’ with indirect efficacy mediated by its propensity to elevate the plasma concentrations of co-administered ASMs. While drug interactions likely contribute to its clinical profile when used as an adjunctive agent, there is also good evidence to suggest that stiripentol possesses antiseizure efficacy in its own right, mediated via type-A ionotropic GABA (GABAA) receptors.39,42
Preclinical and clinical evidence
When administered alone, stiripentol exerts anticonvulsant effects via positive allosteric modulation of the GABAA receptor.39,91 Evidence that stiripentol enhances GABAergic neurotransmission was first provided by experiments conducted using patch-clamp techniques in CA3 pyramidal neurons from the neonatal rat, which demonstrated that, at clinically relevant concentrations, stiripentol enhanced the duration of opening of GABAA receptor channels in a concentration-dependent and barbiturate-like manner. 92 Subsequent studies examined the impact of stiripentol on the functional properties of recombinant GABAA receptors, showing that stiripentol is a positive allosteric modulator of this receptor with greatest effect on receptor complexes containing the α3 subunit. 39 Unlike benzodiazepines, stiripentol is also a strong modulator of δ subunit-containing receptors and does not require the presence of a γ subunit, indicating that its binding site is distinct from some other GABAA receptor modulators.39,42 This unique receptor binding profile is thought to account for the clinical observation that stiripentol is effective in treating childhood seizures, but is less effective in adults, as the α3 subunit is developmentally regulated and shows highest levels of expression in immature brain. 42 Further experiments in a rodent model of pharmaco-resistant status epilepticus have indicated that the subunit selectivity of stiripentol may allow it to remain effective despite seizure-induced changes in GABAA receptor subunit expression during prolonged status epilepticus. 93
Stiripentol was approved for the treatment of seizures associated with Dravet syndrome on the basis of two similarly designed, randomised, placebo-controlled trials, conducted in France and Italy in children with Dravet syndrome aged 3–18 years.94,95 The trials incorporated a 1-month baseline period followed by a 2-month double-blind period in which stiripentol (or placebo) was added to valproate and clobazam.94,95 In both cases, the addition of stiripentol resulted in a significantly greater reduction in the frequency of clonic (or tonic-clonic) seizures, and significantly higher responder and seizure freedom rates, in comparison with placebo.94,95 Mild-to-moderate AEs were common in stiripentol-exposed participants but could be minimised in a sizeable proportion of cases by adjusting comedication dosing.89,94,95 The most frequently reported AEs included drowsiness, slowing of mental function, ataxia, diplopia, loss of appetite resulting in weight loss, nausea and abdominal pain.89,94,95 Sustained efficacy of stiripentol in Dravet syndrome was subsequently demonstrated in a series of open and open-label extension studies; in the open-label extension phase of the original French trial, 57% of patients were classified as responders (⩾50% reduction in seizure frequency) after a mean follow-up duration of 25 months.89,96,97
Cannabidiol
Cannabidiol (C21H30O2) is the primary non-psychoactive cannabinoid of the cannabis plant. 98 Over recent decades, there has been increasing interest in the use of cannabidiol-containing products as potential anti-inflammatory, antiemetic, antipsychotic and antiseizure treatments, but robust evidence for the use of cannabidiol as an ASM has only emerged relatively recently. 44 An initial open-label trial indicated that cannabidiol was sufficiently safe and possessed sufficient efficacy in children and young adults with highly treatment-resistant epilepsy to warrant further investigation. 99 Randomised clinical trials have subsequently been conducted in patients with Dravet syndrome100,101 and LGS.102,103 The European Medicines Agency granted cannabidiol orphan drug status for the treatment of Dravet syndrome in 2014, 104 for the treatment of LGS in 2017 105 and for the treatment of tuberous sclerosis in 2018. 106 Cannabidiol is currently licensed in Europe for use as adjunctive therapy, specifically in conjunction with clobazam, for seizures associated with Dravet syndrome or LGS in patients aged ⩾2 years. 45 In the United States, cannabidiol is licensed for the treatment of seizures associated with Dravet syndrome or LGS in patients aged ⩾1 year. 46 In addition, cannabidiol is licensed for the treatment of seizures associated with tuberous sclerosis complex in Europe (as adjunctive therapy in patients aged ⩾2 years) and the United States (in patients aged ⩾1 year).45,46
Pharmacokinetics and pharmacology
The pharmacokinetic profile of cannabidiol is summarised in Table 1. In addition to having low oral bioavailability (6%), which can lead to significant intra- and inter-individual differences in systemic exposure, 47 it has multiple, complex interactions with a broad range of phase I and phase II hepatic drug metabolising enzymes. In vitro data indicate that cannabidiol is a substrate for CYP3A4, CYP2C19, uridine 5′-diphospho-glucuronosyltransferase (UGT)1A7, UGT1A9 and UGT2B7; an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, UGT1A9 and UGT2B7 at clinically relevant concentrations; and that its active metabolite (7-hydroxy-cannabidiol) is an inhibitor of UGT1A1, UGT1A4 and UGT1A6. 45 Cannabidiol also induces the expression of CYP1A2 and CYP2B6 enzymes at clinically relevant concentrations. 45 This profile is consistent with a drug that is likely to perpetrate and/or endure significant pharmacokinetic interactions. Cannabidiol’s broad-spectrum inhibition of multiple CYP and UGT isoenzymes can be expected to result in elevated plasma concentrations of co-administered drugs, including many ASMs, but relatively few studies have thus far quantified this potential.
There is an important, well-established and bidirectional pharmacokinetic interaction between cannabidiol and clobazam. Co-administration results in an up to fivefold increase in systemic exposure to N-desmethylclobazam, the pharmacologically active metabolite of clobazam, although effects on parent clobazam levels (elevated 20–60%) are much less pronounced.45,107,108 In turn, clobazam elicits modest effects on the plasma concentrations of both cannabidiol (elevated ~30%) and 7-hydroxy-cannabidiol (elevated ~50%).45,107,108 Dose adjustments of cannabidiol and/or clobazam may therefore be required when used together.45,107,108 Likewise, there is also a recognised interaction with stiripentol, which does not appreciably impact cannabidiol concentrations but does decrease the maximum plasma concentration (Cmax) and systemic exposure [area under the time–plasma concentration curve (AUC)] of the active metabolite, 7-hydroxy-cannabidiol, by approximately 30%. 45 Cannabidiol in turn increases the Cmax and AUC of stiripentol by 17% and 30%, respectively, in patients with epilepsy. Interestingly, the initiation of cannabidiol in patients on an established regimen of clobazam and stiripentol does not appear to influence plasma concentrations of clobazam or N-desmethylclobazam, presumably because the hepatic enzymes involved in the metabolism of clobazam (i.e., CYP3A4, CYP2C19) are already maximally inhibited by the presence of stiripentol. 101
The mechanisms of action underlying the antiseizure effects of cannabidiol are currently unclear. Unlike other phytocannabinoids, it appears to have a low affinity for endocannabinoid receptors, CB1 and CB2.98,109 Cannabidiol has instead been proposed to interact with multiple targets involved in the functional modulation of neuronal excitability. These include transient receptor potential vanilloid 1 (TRPV1), the orphan G protein-coupled receptor 55 (GPR55), the equilibrative nucleoside transporter 1 (ENT1) and possibly also voltage-gated sodium channels.110,111 This mechanistic profile is consistent with effects on intracellular calcium signalling, extracellular adenosine concentrations and the vesicular release of neurotransmitters (Figure 3).44,110
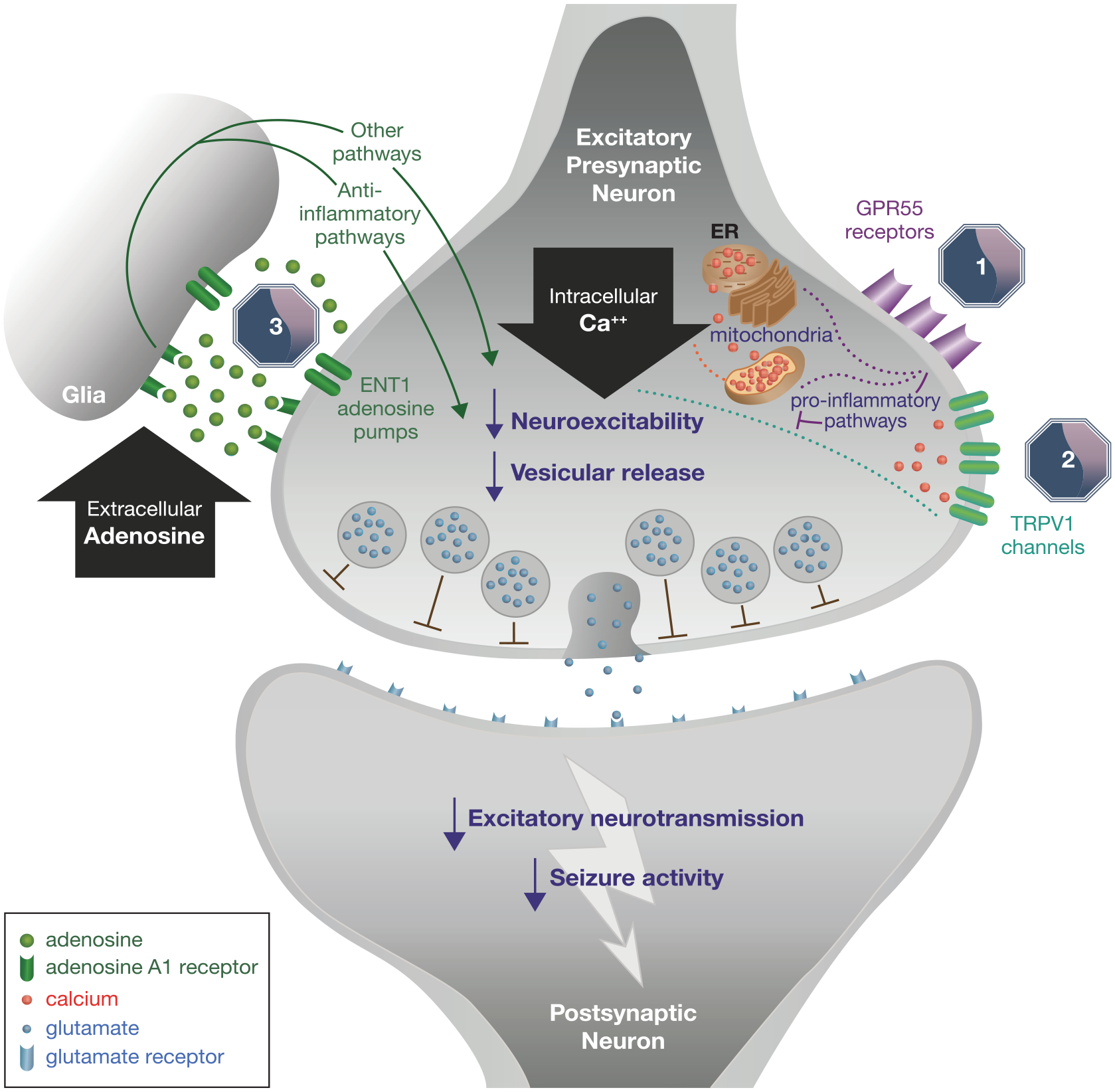
Mechanism of action of cannabidiol.
Preclinical and clinical evidence
Patch-clamp analysis in TRPV1-transfected cells showed that cannabidiol initially activated and then rapidly desensitised TRPV1 in a concentration-dependent manner, effectively blocking the function of these non-selective cation channels. 112 In a related study, cannabidiol was reported to elevate the MES-induced seizure threshold in wildtype but not TRPV1 knockout mice, mechanistically implicating TRPV1 in the anticonvulsive effects of the drug. 113 Cannabidiol is a known antagonist of GPR55, an orphan G protein-coupled receptor (GPCR) that is otherwise activated by a variety of endogenous, phyto- and synthetic cannabinoid compounds.110,114 GPR55 is expressed in both interneurons and principal cells in a range of brain areas involved in seizure generation. Its activation leads to an increase in intracellular calcium ion concentrations, consistent with heightened neuronal excitability, and its expression is also seemingly elevated in epileptic hippocampus.115,116
Further mechanistic studies have shown that intrahypothalamic injection of cannabidiol increases extracellular levels of adenosine in the nucleus accumbens, 117 an effect that is believed to be mediated by inhibition of adenosine reuptake via the ENT1 transporter into presynaptic nerve terminals and adjacent glial cells. 110 Adenosine is a well-characterised endogenous anticonvulsant and the elevation of extracellular adenosine concentrations is a further plausible explanation for the antiseizure activity of cannabidiol. There is also evidence to suggest a role for voltage-gated sodium channels in the mechanistic profile of cannabidiol, with the drug appearing to act as a non-selective inhibitor of recombinant sodium channels, slowing functional recovery from the inactivated state. 111 Other researchers have, however, reported a lack of effect on peak-transient sodium currents and a lack of use-dependent sodium channel block, 110 and questions remain about the validity of this mechanism, given that conventional sodium channel blockers are known to aggravate seizures in Dravet syndrome. 118
Cannabidiol was approved for the treatment of seizures associated with Dravet syndrome on the basis of a double-blind, placebo-controlled trial, in which patients (aged 2–18 years) with Dravet syndrome and drug-resistant seizures were randomised to receive either cannabidiol or placebo, in addition to their standard ASM treatment, for 14 weeks, following a 4-week baseline period. 100 Reductions in the frequency of convulsive seizures and total seizures were significantly greater in patients treated with cannabidiol versus placebo. 100 AEs reported more frequently in patients treated with cannabidiol included diarrhoea, vomiting, fatigue, pyrexia, somnolence and abnormal liver function tests. 100 Cannabidiol’s approval for the treatment of seizures associated with LGS was based on the results of two randomised, double-blind, placebo-controlled trials, in which LGS patients (aged 2–55 years) experiencing ⩾2 drop seizures per week were randomised to receive adjunctive treatment with either cannabidiol or placebo for 14 weeks.102,103 The frequency of drop seizures was significantly lower in the cannabidiol-treated groups, and the most commonly reported AEs included diarrhoea, somnolence, pyrexia, decreased appetite and vomiting.102,103 The longer-term safety/tolerability and sustained efficacy of cannabidiol in the treatment of both Dravet syndrome and LGS have since been demonstrated in a series of open-label extension studies and expanded access programmes.119–122
Ganaxolone
Ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one) is a 3β-methylated synthetic analogue of allopregnanolone, a metabolite of progesterone with antiseizure, neuroprotective and promyelinating properties.123–125 In March 2022, the United States Food and Drug Administration approved ganaxolone for the treatment of seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder in patients aged ⩾2 years. 48 CDKL5 deficiency disorder is a recognised DEE with an estimated incidence of 1 in 42,000 live births. 126
Pharmacokinetics and pharmacology
The pharmacokinetic profile of ganaxolone is summarised in Table 1. Ganaxolone is metabolised by CYP3A4, CYP2B6, CYP2C19 and CYP2D6. 48 CYP inducers decrease ganaxolone exposure, and it is therefore recommended to avoid its concomitant use with strong or moderate CYP3A4 inducers. 48 Patients on a stable ganaxolone dose who are initiating or increasing the doses of enzyme-inducing ASMs (e.g., carbamazepine, phenytoin, phenobarbital and primidone) may need an increase in ganaxolone dose. 48 Like other neurosteroids, ganaxolone acts as a positive allosteric modulator of the GABAA receptor complex, binding to a distinct site from benzodiazepines and increasing the open time of the intrinsic chloride ion pore in response to synaptically released and ambient GABA. 125 Concomitant use of ganaxolone with CNS depressants, including alcohol, may increase the risk of somnolence and sedation. 48
Preclinical and clinical evidence
In preclinical studies, ganaxolone was effective against PTZ-induced clonic convulsions in mice and rats, and demonstrated potent anticonvulsant activity against seizures induced by bicuculline, tert-butylbicyclophosphorothionate (TBPS) and aminophylline in mice. 127 Ganaxolone protects against stage 5 seizures induced by corneal kindling in rats and MES-induced tonic seizures in mice, but, in the latter case, only at doses that produce ataxia on the rotarod test. 127 Ganaxolone also has anticonvulsant activity against flurothyl-induced seizures in rats, 128 and is additionally effective against focal seizures in the 6 Hz model, which is reported to predict drug efficacy in intractable temporal lobe epilepsies. 129 These findings indicate that ganaxolone is a high-affinity, stereoselective, positive allosteric modulator of the GABAA receptor complex that shows potent anticonvulsant activity. 127 More recently, ganaxolone has been shown to promote remyelination in response to gliotoxin-induced focal demyelination of the corpus callosum of ovariectomised rats. 123 The promyelinating properties of ganaxolone were associated with upregulation of key myelin proteins, increased axonal myelination and enhanced microglial-driven clearance of damaged myelin. 123 Preclinical safety, toxicity and mutagenicity studies have been conducted in rodents and dogs. 125 These demonstrated that ganaxolone causes dose-related but reversible sedation, consistent with its GABAergic mode of action. 125
Ganaxolone was approved for the treatment of seizures associated with CDKL5 deficiency disorder on the basis of a phase III trial of adjunctive ganaxolone in children and young adults (aged 2–21 years) with CDKL5 deficiency disorder (NCT03572933). 130 Following a 6-week prospective baseline period, patients were randomised (1:1) to receive treatment with either adjunctive ganaxolone or matching placebo for 17 weeks. 130 The median percentage reduction in 28-day motor seizure frequency was significantly greater for ganaxolone versus placebo (30.7% versus 6.9%). 130 The most commonly reported treatment-emergent AEs (reported by ⩾10% of patients, higher incidence in ganaxolone versus placebo groups) were somnolence, pyrexia and upper respiratory tract infections. 130
A review of initial human experience with ganaxolone indicated that most AEs were mild (82%) or moderate (14%) in intensity and predominantly limited to headache, dizziness, somnolence, gastrointestinal disturbances and malaise. 131 Ganaxolone has demonstrated promising efficacy as adjunctive therapy in an open-label, dose-escalation study in paediatric patients (aged 7 months to 7 years) with refractory West syndrome, 132 and in an open-label study of children (aged 2–15 years) with LGS. 133
ASMs in development for DEEs
Other novel and repurposed ASMs currently in clinical development for use in DEEs include soticlestat, carisbamate, verapamil, radiprodil, clemizole and lorcaserin (Table 2).134–137
Summary of current clinical information for ASMs in development for DEEs.

AE, adverse event; ASM, antiseizure medication; DEE, developmental and epileptic encephalopathy; 24HC, 24S-hydroxycholesterol; LGS, Lennox–Gastaut syndrome; OLE, open-label extension; PCDH19, protocadherin 19; RCT, randomised controlled trial; TEAE, treatment-emergent adverse event.
Soticlestat
Soticlestat (TAK-935/OV935; [4-benzyl-4-hydroxypiperidin-1-yl][2,4′-bipyridin-3-yl] methanone) is a novel, first-in-class small molecule that selectively inhibits cholesterol 24-hydroxylase (CH24H), a brain-specific enzyme that converts cholesterol into 24S-hydroxycholesterol (24HC), an endogenous positive allosteric modulator of the N-methyl-D-aspartate (NMDA) subtype of glutamate receptor.138–140 In preclinical studies, soticlestat reduced brain 24HC in a dose-dependent manner, suppressed potassium-evoked extracellular glutamate elevations in the hippocampus, and protected against premature death in APP/PS1-Tg mice (a transgenic model carrying human amyloid precursor protein and presenilin-1) at a dose associated with a 50% reduction in brain 24HC. 138 Although reported to be ineffective in a range of traditional seizure models, soticlestat has been shown to prevent audiogenic seizures in Frings mice and to delay the development of generalised seizures in the rat amygdala kindling model. 141 Soticlestat’s novel mechanism of action is thought to confer the potential to reduce seizure frequency and severity, particularly in disorders associated with neuronal hyperexcitability and enhanced glutamatergic transmission.134,138
The pharmacokinetic profile, genotoxicity and toxicology of soticlestat have been investigated successfully in rats and dogs, leading to phase I assessment in 48 healthy human subjects. 139 Soticlestat was well tolerated up to a single oral dose of 1350 mg. 139 AEs were mild and did not appear to be dose-related; the most frequently reported AEs were headache, electrocardiogram electrode application-site dermatitis and nausea. 139 When administered via oral solution, soticlestat was rapidly absorbed [median time to maximum plasma concentration (tmax), 0.25–0.52 h), mean terminal elimination half-life (t½) ranged from 0.8 to 7.2 h and mean volume of distribution (Vz/F) ranged from 732 to 2240 l across doses. 139 Administration of soticlestat in tablet form, or with food, lowered maximum plasma concentration (Cmax) and delayed tmax but did not affect overall exposure. 139 Plasma 24HC concentrations decreased with increasing soticlestat dose, with a maximum mean decrease of 23% at 16 h after the 900 mg dose. 139 Soticlestat has been assessed as adjunctive therapy in a phase II, multicentre, randomised, double-blind, placebo-controlled trial in paediatric patients (aged 2–17 years) with Dravet syndrome and LGS (the ELEKTRA study) 135 and in a phase Ib/IIa study in adults with DEEs (NCT03166215; Table 2). 134 A phase II open-label extension study (NCT03635073) of adjunctive soticlestat in children and adults with DEEs, and phase III trials of soticlestat as adjunctive therapy in children and young adults with Dravet syndrome (NCT04940624) and children, teenagers and adults with LGS (NCT04938427), are currently ongoing.
Carisbamate
Carisbamate (RWJ-333369; S-2-O-carbamoyl-1-o-chlorophenyl-ethanol) belongs to the alkyl-carbamate family of drugs, which also includes cenobamate and felbamate.142–144 Preclinical studies have shown that carisbamate has broad-spectrum antiseizure and potentially antiepileptogenic effects in a range of models of focal, generalised and limbic epilepsy at doses below those that produce CNS toxicity.145–148 It has also been shown to be effective in a rat model of symptomatic infantile spasms that might indicate efficacy in West syndrome. 142 The mechanism of action of carisbamate appears to be multi-factorial, involving blockade of voltage-gated sodium channels, T-type calcium channels and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- and NMDA-mediated excitatory neurotransmission.149,150 There is also evidence indicating possible potentiation of chloride conductance via GABAA receptors, but this is not well characterised at present. 151 In healthy human subjects, oral doses of carisbamate are well absorbed (>95% of total dose), and tmax is 1–3 h in the fasted state.152,153 The apparent volume of distribution is approximately 50 l (indicating non-extensive peripheral distribution), and plasma protein binding is approximately 44%.152,153 Around 94% of the administered dose is recovered in urine, with just 2% recovered as unchanged carisbamate.152,153 Available evidence suggests that the most frequently reported AEs appear to be CNS-related, generally mild and transient, and include headache, dizziness, somnolence and nausea.152,153 Phase I studies of carisbamate in adult and paediatric patients with LGS (NCT03731715, NCT04062981) are currently ongoing.
Verapamil
Verapamil (2-(3,4-dimethoxyphenyl)−5-[2-(3,4-dimethoxyphenyl)ethyl)-methylamino]−2-propan-2-ylpentanenitrile;hydrochloride) is a voltage-gated calcium channel blocker from the phenylalkylamine class that is licensed in Europe for the management of hypertension, angina pectoris, paroxysmal supraventricular tachycardia and atrial fibrillation.154,155 Verapamil shortens epileptic depolarisations and reversibly blocks transmembrane calcium conductance during epileptic discharges, resulting in a dampening of seizure-like activity.156,157 In addition to its calcium channel-blocking properties, it acts at the blood-brain-barrier to inhibit P-glycoprotein [Pgp; multidrug resistance protein 1 (MDR1)], a drug efflux transporter that has been suggested to limit brain access of several ASMs.158,159 It is also a recognised inhibitor of the drug metabolising enzyme CYP3A4 and has a known pharmacokinetic interaction with carbamazepine. 160 As such, verapamil might be expected to elicit antiseizure effects via both direct (i.e., reduced calcium conductance) and indirect (i.e., enhanced plasma concentrations and/or brain penetration of concomitantly administered drugs) mechanisms.
Verapamil is more than 90% absorbed after oral administration, its plasma protein binding approaches 90%, and its volume of distribution is 1.8–6.8 l/kg in healthy subjects. 154 Steady state is reached 3–4 days after multiple once daily dosing, with a t½ of 3–7 h. 154 Approximately one-half of an administered dose is eliminated renally within 24 h, 70% within 5 days, and 3–4% is excreted renally as unchanged drug; in addition, up to 16% is excreted in faeces. 154 Although there is some evidence to support the use of verapamil in intractable epilepsies, 161 by virtue of its inhibitory effect on Pgp, a carefully designed study of the efficacy and tolerability of adjunctive verapamil in a canine model of phenobarbital-resistant epilepsy showed dose-limiting adverse effects on cardiovascular function and was discontinued early due to deterioration in seizure control in some animals. 162 Clinical data on the use of adjunctive verapamil in the treatment of DEEs are currently limited to small case series, including a pilot study that included four patients with Dravet syndrome and one patient with LGS.159,163 Another phase II study in children and young adults with Dravet syndrome has recently been completed (NCT01607073), but no data are yet available (Table 2). It is also worthy of note that verapamil showed pronounced efficacy against seizures associated with LGS, in what is arguably the landmark case of precision medicine in DEEs. 164
Radiprodil
Radiprodil (RGH-896; UCB3491; N-(2,3-dihydro-2-oxo-6-benzoxazolyl)-4-[(4-fluorophenyl)methyl]-alpha-oxo-1-piperidineacetamide) is a negative allosteric modulator of the NMDA subtype of glutamate receptor, with selectivity for receptor complexes containing the GluN2B subunit.165,166 Gain-of-function mutations in the gene encoding this subunit (GRIN2B) have been linked with several DEEs, including West syndrome. 166 Radiprodil has demonstrated dose-dependent efficacy against generalised clonic convulsions in audiogenic seizure-sensitive mice and generalised tonic seizures induced by PTZ in immature Wistar rats but was without effect against focal seizures in the 6 Hz model and PTZ-induced tonic seizures in adult rats. 136 In healthy human subjects, the median tmax of radiprodil is 4.0 h (range 3.0–6.0 h), the geometric mean t½ is 15.8 h and the apparent volume of distribution is 334 l. 165 Radiprodil has shown preliminary efficacy as add-on treatment in a phase Ib open-label, ascending dose study in three infants with West syndrome (aged 5–11 months) who were resistant to combination therapy with vigabatrin and prednisolone 136 (Table 2). The most common AEs in this small-scale study were vomiting and pyrexia, neither of which merited discontinuation, but the study was ultimately terminated due to insufficient enrolment. 136
Clemizole
Clemizole (1-[(4-chlorophenyl)methyl]-2-(pyrrolidin-1-ylmethyl]benzimidazole)hydrochloride) is a first-generation antihistamine drug, with selectivity for the H1 subtype of histamine receptor. It is also an intracellular inhibitor of hepatitis C virus protein synthesis and was originally developed and used as an antiviral drug in the 1950–1960s.15,167 Experiments conducted in scn1Lab mutant zebrafish larvae (an experimental model for Dravet syndrome) have shown that clemizole can attenuate behavioural and electrographic seizure activity, but that these effects are mediated via modulation of serotonergic rather than histaminergic signalling.167,168 Clemizole is rapidly metabolised in mice with a plasma half-life of less than 10 min (compared to 3.4 h in humans), hampering its evaluation in murine seizure and epilepsy models. 167 Clemizole is orally bioavailable and its metabolism is mediated via CYP3A4, but further information on its pharmacokinetic profile in humans is currently lacking. 15 In the absence of clinical grade clemizole for use in human trials, a recent study instead explored the efficacy of lorcaserin, a clinically approved 5-HT2C receptor agonist identified from screening of compound libraries for drugs that could recapitulate the antiseizure activity of clemizole in scn1Lab zebrafish mutants. 167 Five children with medically intractable Dravet syndrome (aged 7–18 years) were treated with lorcaserin as part of a compassionate use off-label programme, with all five experiencing a reduction in the total number of seizures and three with significant reductions (up to 90%) in generalised tonic-clonic seizures. 167 The most common AE was decreased appetite, but otherwise lorcaserin was well tolerated, and there were no discontinuations due to side effects. 167 These findings provide a rationale for further investigation of clemizole (and/or other 5-HT2 agonists) as a potential treatment for Dravet syndrome. Indeed, a phase II study of adjunctive clemizole therapy to control convulsive seizures in patients with Dravet syndrome is currently ongoing (NCT04462770) (Table 2).
Lorcaserin
Lorcaserin ([R]−8-chloro-1-methyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride hemihydrate) is a selective serotonin receptor (5-HT2C) agonist that was approved in the United States for the management of obesity in 2012, but subsequently withdrawn due to evidence of an elevated risk of cancer following long-term use.137,169,170 Lorcaserin has been shown to have anticonvulsant properties in an experimental model of limbic seizures, 171 and to suppress absence seizures in the Genetic Absence Epilepsy Rats from Strasbourg (GAERS) model in a dose-dependent manner. 172 In healthy adults at steady state, the tmax for the immediate release formulation of lorcaserin is 1.5 h and the t½ is approximately 12 h. 173 Lorcaserin distributes to the cerebrospinal fluid and CNS in humans and is moderately bound to human plasma proteins (~70%). 170 It is extensively metabolised in the liver by multiple enzymatic pathways and its metabolites are excreted renally. 170 Lorcaserin is also an inhibitor of CYP2D6. 170
In addition to the pilot study of lorcaserin in five patients with Dravet syndrome reported above, 167 a subsequent retrospective case series of patients with Dravet syndrome (n = 20), LGS (n = 9) and other treatment-resistant focal and generalised epilepsies (n = 6) demonstrated a significant reduction in the frequency of motor seizures following treatment with lorcaserin (Table 2). 137 The most common AE was again decreased appetite (19.6% of patients), followed by decreased attentiveness (17.1%) and weight loss (11.1%). 137 Five patients discontinued lorcaserin within the first 30 days as a result of side effects. 137 A phase III trial of adjunctive lorcaserin therapy in patients with Dravet syndrome (NCT04572243), and an extended access programme for the treatment of Dravet syndrome and other refractory epilepsies (NCT04457687), are currently ongoing (Table 2).
Summary and conclusions
The past 15 years have witnessed almost as many approvals of new ASMs for DEEs as have been introduced for common epilepsies, with five drugs (i.e., rufinamide, fenfluramine, stiripentol, cannabidiol and ganaxolone) licensed specifically for the treatment of Dravet syndrome, LGS, CDKL5 deficiency disorder and/or tuberous sclerosis complex. The focus on drug development for DEEs meets an extremely high unmet need in this group of rare neurodevelopmental disorders that are typically characterised by early-onset and often intractable seizures, together with developmental delay/regression, electroencephalographic abnormalities and elevated incidence of SUDEP.4,5,14,15 These advances can be explained, at least in part, by a greatly improved understanding of the aetiology of the DEEs, advances in genetic methodologies and in the coordination of international epilepsy genetics studies, and by the efforts of the ILAE and expert investigators to standardise phenotypic classification of these conditions.2,9,174 In addition, the opportunities afforded by orphan drug designation, which expedites clinical drug development in rare diseases, should not be overlooked. 175 Surgical and dietary interventions have also emerged, and gene therapies are in development, but drug therapy remains the cornerstone therapeutic approach for the management of seizures in patients with DEEs.
Mechanisms of action amongst approved treatments for DEEs are diverse, encompassing what might be considered both conventional and novel ASM targets. Rufinamide is just one of many sodium channel-blocking drugs used in epilepsy and that traditionally possess efficacy against focal and generalised tonic-clonic seizures. Many of those drugs, for example, carbamazepine, are either ineffective or can exacerbate seizures in DEEs,18,19,176 so the efficacy of rufinamide in LGS is somewhat perplexing. Like other sodium blockers, it is believed to interfere with the functional recycling of sodium channels following depolarisation, slowing the recovery of channels from the inactivated state back to the resting state and thereby limiting the propensity of individual neurons to fire at high frequency.33,54,55 It is possible that the additional effect of rufinamide on the activation, rather than just inactivation, of sodium channels is what sets this drug apart. 56 Stiripentol and ganaxolone also seemingly possess conventional mechanisms of action, acting as positive allosteric modulators of the GABAA receptor complex and enhancing cellular responses to the inhibitory neurotransmitter GABA.39,91,127 In this regard, they behave in a similar manner to barbiturates and benzodiazepines, with closer pharmacological profiles to the former. Activity of stiripentol and ganaxolone at all GABAA receptor isoforms, including those that contain the δ subunit, and their effects on chloride channel kinetics (enhancing duration of channel opening), suggest a barbiturate-like action at both synaptic and extra-synaptic receptor sites,39,91,92,127,177 and distinguish these drugs from benzodiazepines that increase frequency of channel opening and are devoid of efficacy at δ-containing receptors. The apparently preferential efficacy of stiripentol at receptor complexes containing α3 subunits may also be important, not only in terms of distinguishing it from barbiturate drugs but also in underpinning its efficacy in DEEs, given the developmental regulation of this receptor subunit and the reported resistance of its expression to prolonged seizure activity. 39
In addition to conventional ASM targets, two of the recently approved treatments for seizures associated with DEEs appear to possess novel pharmacological mechanisms. On the basis of available evidence, fenfluramine appears to exert its antiseizure effects by a combined action on serotonergic transmission and the sigma-1 receptor.37,71–75 The clinical profile of fenfluramine as an appetite suppressant is consistent with a serotonergic mechanism, as are its range of adverse effects and its pharmacodynamic interaction with antidepressant agents. It would appear that fenfluramine can release serotonin from vesicular storage, leading to an elevation in extracellular serotonin concentrations and consequent activation of a range of 5-HT receptors. 71 Of those receptors, the 5-HT2 family is likely of greatest prominence, and this is reinforced by the fact that two of the putative ASMs in current development for DEEs, namely clemizole and lorcaserin, also target this receptor subtype. The effects of fenfluramine on sigma-1 receptors are less well characterised, but there is reasonable evidence, at least at the preclinical level, of their contribution to the anticonvulsant actions of the drug and plausibility in its apparent interaction with endogenous neurosteroids. 75 The pharmacology of cannabidiol remains an enigma; it does not appear to interact with cannabinoid receptors but instead to influence a range of cellular processes that can alter neuronal excitability. TRPV1 is a novel target amongst ASMs and more commonly recognised as a modulator of pain signalling in the peripheral nervous system, 178 but the loss of anticonvulsant efficacy of cannabidiol in TRPV1 knockout animals is suggestive of an additional central role. 113 GPR55 and ENT1 are further novel targets by which cannabidiol has been proposed to act, each of which is again credible but requires further investigation. 110 Voltage-gated sodium channels have also been implicated in the mechanism of action of cannabidiol and there is some evidence to support a non-selective inhibitory effect on channel function. 111 It is also possible, however, that this accounts not for the beneficial effects of the drug but for the occasional seizure worsening that was observed in clinical trials of cannabidiol in Dravet syndrome and LGS, particularly in patients who were not additionally taking GABAergic medications. 179
In addition to approved ASMs for DEEs, a broad range of treatments with a hugely diverse pharmacology remain in clinical development. Some of these have been evaluated previously in common epilepsies (e.g., carisbamate), some have been repurposed from other therapeutic areas (e.g., verapamil, clemizole, lorcaserin), and others are new small molecules specifically designed for use in CNS disorders (e.g., soticlestat, radiprodil). Their cellular effects transcend conventional antiseizure drug mechanisms, such as blockade of voltage-gated sodium and calcium channels and inhibition of glutamate receptors, but also include novel targets, including the serotonergic system and brain cholesterol metabolism. All have demonstrated evidence of anticonvulsant efficacy at the preclinical level,136,141,147,148,156,157,167,171,172 and some have shown beneficial effects in early phase trials,134–137,163,167 but, at this time, it is not clear whether these drugs will ultimately reach the marketplace.
Taken together, the approved and investigational drugs for DEEs highlight some important advances in ASM pharmacology. Repurposing of already approved treatments is prominent, partly because the existence of known toxicology profiles allows a more rapid transition to clinical testing but also because it provides an opportunity to deploy existing drugs with known mechanisms of action in a targeted or precision manner in the light of new aetiological information.180–182 Although still in development, soticlestat is also arguably repurposed, possessing a mechanism that might more obviously be associated with the treatment of dementias rather than epilepsy. 140 Several drugs appear to act as allosteric modulators of receptor function (positive or negative) as opposed to being full agonists or antagonists, and some additionally have a narrow selectivity for receptor subtypes and/or subunits, both of which might offer a more refined approach to manipulating neuronal excitability, potentially providing efficacy but limiting wider adverse consequences. Another interesting development is the (re)-emergence of serotonin as a drug target in the treatment of seizures. Lowering of the seizure threshold with antidepressant drugs has coloured the view of serotonin and serotonergic agents in epilepsy for several decades,183,184 despite experimental evidence supporting a role for this neurotransmitter in seizure regulation and in the pharmacology of some traditional ASMs.185–187
That said, there remains considerable uncertainty about the precise mechanisms of action of the drugs described in this article, not least cannabidiol. Much of the research reported on these drugs to date has been relatively limited in scope and has often employed reductive models and methods, with mechanisms often deduced by a process of elimination using pharmacological tools of unclear selectivity. Much more effort is required to determine whether effects on TRPV1, GPR55, serotonergic transmission, cholesterol metabolism, etc. are genuinely responsible for reported anticonvulsant efficacy.
Although the approved ASMs for DEEs rarely render patients seizure free, their efficacy is otherwise beyond doubt and that of emerging drugs is similarly promising, although whether this proves to be sufficiently robust in larger scale trials remains to be seen, especially given the inherent intractability of these conditions.9,14,15,17 The low prevalence of DEEs, which underpins the orphan drug designation through which many of these ASMs have been developed, makes the unequivocal identification of efficacy challenging and necessitates collaborative efforts in phenotype characterisation, trial design and recruitment, and in the use of standardised trial endpoints. 188 In addition, several agents present challenging pharmacokinetic issues, with the potential to perpetrate and/or endure extensive drug-drug interactions that are confounded by the routine use of polypharmacy in the treatment of DEEs. Others have been associated with long-term AEs that previously warranted their withdrawal from the market, although, in the case of fenfluramine at least, this appears to be ameliorated through the use of lower drug doses. 189 These drugs also have distinct safety profiles, particularly with regard to psychobehavioural AEs and effects on sleep and cognition, which can be either positive or negative, and which therefore require careful consideration when making treatment decisions. 190
The striking feature of approved and emerging ASMs for DEEs is their pharmacological diversity, encompassing both conventional and novel drug targets and a degree of mechanistic subtlety not previously seen, which has expanded our understanding of what might reasonably constitute an antiseizure mechanism. This diversity gives hope to the alleviation of not only seizures but also other disabling symptoms of these conditions. The future is brighter now for DEEs than it was just 10–15 years ago; we now have drugs at our disposal to better treat a range of specific DEEs, including Dravet syndrome, LGS and CDKL5 deficiency disorder, and yet more drugs on the horizon. Further understanding of the aetiologies of DEEs and continued improvements in phenotype characterisation and in genetic analysis should enable the development or repurposing of yet more molecularly targeted ASMs.