
Abstract
Colorectal cancer (CRC) is a highly prevalent disease, and despite advances in medical research, much remains unknown about CRC. As such, it is important to improve our current understanding of CRC. Several animal models for CRC exist, and they provide an excellent tool for studying CRC tumorigenesis. These models, however, have limitations, and a good understanding of the pathophysiology of these models is required in order to fully understand how closely they mimic human sporadic and colitis-associated CRC.
Introduction
According to the International Agency for Research on Cancer, colorectal cancer (CRC) was the third most common cancer in men and the second in women worldwide with an estimated 694,000 deaths in 2012. 1 Inflammatory bowel disease (IBD), Crohn's disease (CD), and ulcerative colitis (UC) are chronic relapsing inflammatory disorders of unknown etiology that result in mucosal ulceration and macroscopic inflammation of the gastrointestinal tract. UC affects only the colon, while CD may affect any part of the gastrointestinal tract, but is most likely to involve the terminal ileum and colon. 2 IBD increases the risk of CRC, and the risk appears to be associated with the length of time since the disease was diagnosed, the extent of the colon that is subjected to the chronic inflammation, and how well the inflammation is kept under control. 3 Despite advances in medical research, however, much of CRC pathogenesis, both sporadic and inflammation-associated, remains unclear.
To improve the detection of CRC and the allocation of treatment, our current understanding of CRC needs to be improved. Studying the process of malignant change in humans, however, is difficult due to the medical treatment of the patients and the inability to trial drugs in the prodromal phases of CRC development. On the other hand, animal models provide the flexibility of creating a model that suits the research question of interest, with large and reproducible numbers of animals. Most importantly, it allows researchers to study tumor progression. These methods allow new insights that could lead to a better management of disease detection and treatment.
For CRC, as with many other diseases, multiple animal models exist, with the differences between each model potentially allowing the researcher to focus on one aspect of disease progression. This review, therefore, aims to summarize several murine models that relate to IBD-associated and sporadic CRC, and to discuss how these models impact on our current understanding of CRC.
Animal Models of IBD-Associated Colorectal Cancer
The method used to induce colonic tumors depends on research questions such as studying the adenoma–carcinoma pathway of sporadic tumorigenesis or an alternate tumor model such as serrated colorectal polyposis. As such, there are a variety of colorectal tumor-inducing regimes. These can be grouped under transgenic, chemically induced, and diet-based models, and will be discussed below.
Transgenic models
Transgenic models are useful for studying the effect of a particular gene in CRC development, and the section below will describe the use of the APCMin model, Smad mutant mice, and the creation of conditional transgenic animals using Cre-lox recombination and Rag2 deficiency for the development of spontaneous CRC.
Mouse models harboring a mutated Apc gene
Germline mutations occur in the adenomatous polyposis coli (APC) gene located on chromosome 5q21 and often lead to protein truncation, which is responsible for the phenotype of familial adenomatous polyposis (FAP). Also, biallelic mutations to the APC gene are often detected in early sporadic adenomas.4,5 APC consists of 15 coding exons and several noncoding exons, and alternate splicing results in several isoforms. 6 APC encodes a 2845-aa (amino acid) protein, 7 which functions as a scaffold protein that affects both cell adhesion and migration and is part of a signaling pathway complex mediated by the Wnt pathway. 6 This pathway regulates both the phosphorylation and degradation of β-catenin, which is an intracellular protein that binds E-cadherin and links it to the actin cytoskeleton and phosphorylation of β-catenin. This results in the ubiquitinylation of β-catenin and its subsequent degradation. Mutations in APC, therefore, lead to cytoplasmic accumulation of β-catenin, allowing it to bind to the T-cell family of transcription factors affecting expression of genes related to cellular differentiation, proliferation, migration, and apoptosis, such as cyclin D-1 and c-Myc oncogene. 6 There are several mouse models of CRC that involves mutations to Apc, such as the ApcMin, Apcδ716/+, and Apc1638 transgenic mice.
The multiple intestinal neoplasia (Min) allele in the Apc gene was discovered in an ethylnitrosourea mutagenesis assay, and mice harboring this allele are referred to as APCMin mice. 8 This allele is inherited in an autosomal dominant fashion and features a nonsense mutation in codon 850 giving rise to a truncated APC protein.7,8 The mutation is maintained by crossing Min/+ males with C57Bl/6J female mice, affecting 50% of the progeny. Spontaneous development of multiple colonic adenomas occurs throughout the large and small intestine, with locally invasive tumors forming in aged mice. 8 Anemia is a secondary characteristic of ApcMin mice. Due to the spontaneous development of multiple tumors and a germline mutation in Apc that is similar in FAP and sporadic CRC patients, this model is commonly used to study sporadic CRC and FAP. 9
The Apc gene in Apcδ716 mice is truncated at codon 716, and mice heterozygous for the mutation developed multiple polyps in the intestinal tract shortly after birth. 10 In the same study, it was shown that polyp development continued with age, and by 7 weeks after birth all mice developed polyps. These polyps varied in size and stage, and were found from the duodenum to the rectum, with most polyps occurring in the small intestine. Histologically, the polyps were polypoid, papillary, or sessile adenomas, and most tumors remained in the lamina propria. Also, loss of heterozygosity of Apc was found in all nascent microadenomas, suggesting that, in this model, loss of heterozygosity is one of the earliest changes in microadenoma formation. Interestingly, there were more tumors in Apcδ716/+ than in ApcMin mice despite both models using C57Bl/6J mice, suggesting that the extra 134 aa in ApcMin mice might be responsible for this difference.
In Apc1638 transgenic mice, a mutation to aa 1638 was achieved by the insertion of a neomycin phosphotransferase expression cassette into the Sma I site corresponding the codon 1638 of the Apc cDNA. This resulted in a 1660-aa truncated protein, of which the first 1637 aa correspond to that of normal Apc. 11 In this study, at 10 weeks of age, chimeric and heterozygous mice exhibited colonic polypoid hyperplastic lesions. These lesions featured small spherical elevation with or without a stalk presenting on the mucosal crest, active proliferation as evident by the elongation of colonic crypts, and an increased number of mitotic figures reaching a high position in the crypt columns. In 20-week-old mice, the animals also developed tumors of the small intestine, which were both benign adenomas and malignant adenocarcinomas and mostly found in the duodenum and jejunum. Tumor size varied, and histological analysis revealed that the adenomas were either villous or tubulovillous with severed dysplasia. On the other hand, adenocarcinomas displayed high–to-moderate differentiation with infiltration into the muscularis mucosa, submucosa, or inner layer of the muscularis. Seventy percent of mice did reach 12 months of age, and in two mice that did, tumors were located in the colon, stomach, and small intestine. Despite sharing several phenotypes, Apc1638/+ mice appear to survive longer than ApcMin mice, suggesting that this difference might be a result of the difference in 788 aa.
Therefore, a greater understanding of the function of Apc can be achieved by comparing the tumors between these different Apc mutant models, and may serve as a platform for better CRC therapy.
Smad mutant mice
Smad proteins are downstream signal transducers of the transforming growth factor-β (TGF-β) signaling pathway and are involved in the regulation of cell growth (Fig. 1). Loss-of-function mutations to genes such as Smad 2 and Smad 4 have been found in CRC patients. Compared to SMAD 2, 3, 6, and 7 genes, higher frequencies of SMAD 4 gene mutations are found in CRC patients with distant metastasis. 12 Mice that are haploinsufficient for Smad 4, a key TGF-β signal transducer, develop serrated adenomas and mixed polyposis throughout the upper gastrointestinal tract. 13
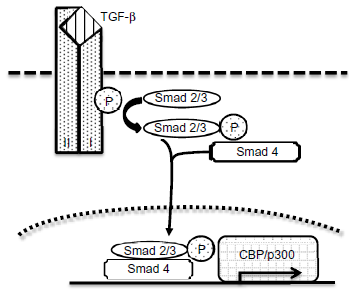
An overview of TGF-β and Smad signaling activation. At the cell surface, binding of the TGF-β ligand to transmembrane receptor serine/threonine kinases (types I and II) induces the activation of Smad 2/3 proteins via phosphorylation. Activated Smad 2/3 proteins forms a complex with Smad 4 and translocates into the nucleus where it regulates the transcription of target genes via the binding protein of cAMP response element binding protein (CBP) or p300 coactivators or repressor.
Microscopic examination of mice that are haploinsufficient in Smad 4 did not reveal any neoplastic lesions at 6 months of age, and colorectal polyps were found only in 1 out of 10 mice at age 15 months, and in 2 out of the remaining 9 mice at 18 months, confirming the late-onset nature of tumor phenotype of this model. 13 Histopathological analysis of these tumors confirmed that polyps were hyperplastic with branching villi and were occasionally serrated.
As invasive CRC can originate from both serrated and hyperplastic polyps, and different cancer pathways are thought to contribute to different tumor morphologies,14,15 this model presents an opportunity to study carcinogenesis based on alternate tumorigenesis pathways.
Smad 3 knockout mice are noted to develop intestinal adenocarcinomas that are independent of Apc mutations, and cancer is also detected in the intestinal draining lymph nodes. 16 This model of metastasis highlights the tumor-suppressive role of Smad 3 in colonic epithelium. As it presents multiple tumor morphologies and cancer stages that are similar to those seen in humans, it may also be useful in studying colorectal tumorigenesis.
Takaku et al developed a model where mice containing mutant Smad 4 were cross-bred with Apcδ716 mice, resulting in compound mutant mice of both Smad 4 and Apc. 17 At 14 weeks, the small intestinal polyps of the compound mutant mice were larger than those of Apcδ716 mice, and similarly, some colonic polyps in the compound mice at weeks 14 and 20 of age developed into larger sizes that were not observed in Apcδ716 mice. Despite developing larger polyps, compound mutant mice developed fewer polyps than Apcδ716 mice. At 14 weeks of age, compound mutant mice had 82 polyps compared to 676 in Apcδ716 mice. It is of interest to note that the compound mutant mice developed adenocarcinomas with extensive stromal invasion, whereas Apcδ716 mice developed tumors that were mostly in the lamina propria. 10 This demonstrates that Smad 4 mutations are sufficient in causing malignant transformation of Apcδ716 polyps.
Cre-lox-targeted gene deletion
The Cre-lox recombination system can be used for the specific deletion of genes with the genetic sequence to be deleted flanked on either end by LoxP sites. In the presence of Cre recombinase, a cut is made through the core sequences, and inverted repeats on either side of the target sequence are spliced. This creates a circularized target sequence containing a LoxP site, which is excised from the genome.
The benefit of using the Cre-lox system is that the conditional expression of Cre recombinase allows a Cre-lox deletion to occur only within a specific organ, such as the intestine, as demonstrated by the creation of mice expressing a β-catenin mutation solely in the intestine. Using this method, exon 3 of the β-catenin gene was deleted in the intestine and, when examining mice heterozygous for this mutation, polyposis was present in the small intestine but not in the cecum or colon, although nascent microadenomas were found in the colonic mucosa. 18 The observation suggests that colonic tumorigenesis can thus develop through the Wnt signaling pathway.
The Cre-lox deletion system was also used to induce a conditional loss of Apc in mice. 19 A conditional Apc knockout model, Apclox/lox, in which exon 14 of Apc was flanked by LoxP sites, was crossbred with transgenic mice that expressed tamoxifen-dependent Cre recombinase conditionally and specifically in the intestinal epithelium. Four days after one injection of tamoxifen into mice resulted in the moribund phenotype of mutant mice. Histological studies of the small intestine identified features of dysplasia such as nuclear pseudostratification, enlargement of the nuclei, prominent nucleoli, and basophil accumulation. Similar studies of the colon revealed less drastic features. The small intestine featured an enlargement of the crypts due to enhanced cell proliferation, induction of apoptosis, and impairment of migration. A lack in epithelial stem cell differentiation was also noted, and high β-catenin expression was present in highly proliferative areas. This again demonstrates that Apc is important in maintaining intestinal homeostasis and that disruption of this gene may contribute to the development of CRC.
It is of interest to know that, in addition to the conditional Cre-lox deletion system, there is a model of temporal gene expression regulation using the Cre-lox deletion system. In such a system, the expression of the target gene can be regulated by experimental conditions such as cytokine expression within a tumor and changing levels of growth factors. For example, a tetracycline-responsive regulatory system was tested for its ability to temporally control gene expression in transgenic mice. 20 In this system, a fusion of tetracycline controlled trans-activator protein (tTA) was used, which was composed of the tetracycline-resistant operon repressor tet and the activating domain of viral protein VP16 of the herpes simplex virus. tTA strongly activates transcription from PhCMV*-1, a minimal promoter from human cytomegalovirus (hCMV) fused to tet operating sequences. iTA binds to the tet operator sequences in the absence of tetracycline but not in its presence, resulting in reduced transcription in the presence of tetracycline. Using this system, the authors were able to 1) induce the expression levels of the reporter genes luciferase and β-galactosidase by several thousand-fold and 2) reduce their levels back to baseline with the administration of tetracycline.
Therefore, this system might be useful in temporally controlling the expression of genes involved in pro-tumorigenic processes to further define their roles normal growth and tumorigenesis.
Rag2-deficient mice
B and T lymphocytes are unable to mature in mice that are deficient in the recombinase-activating gene (Rag) 2, and these mice are noted to develop colitis-associated CRC 4 months after being infected with Helicobacter hepaticus. 21 By contrast, H. hepaticus-infected wild-type mice displayed minimal colitis and did not develop CRC. In addition, when an adoptive transfer of purified CD4+ CD45RBlo CD25+ T-cells occurred 72 hours before the H. hepaticus infection, development of colitis and hyperplasia was significantly reduced. In 7 of 17 mice that received adoptive transfer of purified T cells prior to infection with H. hepaticus, no evidence of colitis or dysplasia of the colon was observed, while only 4 of the mice experienced minimal or mild inflammation. The remaining six mice, however, developed localized moderate or severe inflammation and epithelial dysplasia, thus demonstrating the importance of lymphocytes in the protection against CRC development. It should also be noted that the progression of IBD-associated epithelial dysplasia and carcinoma in this model resembled the dysplasia sequence observed in the IBD-associated CRC development in humans. 22
Kirsten-ras mutant models
According to Vaughn et al, about 50% of CRC tumors contained mutations to Kirsten (K)-ras. 23 K-ras mutations tend to be observed in the early phase of IBD-associated colorectal tumorigenesis and in the later phases of sporadic CRC.5,24,25 Due to the prevalence of K-ras mutations in CRC tumors, it is of importance to understand its function in CRC tumorigenesis. To achieve this, CRC was studied in mice transgenic for a K-ras with a glycine to valine mutation at codon 12 (K-rasV12G). 26 In this study, 31 out of 41 mice developed intestinal tumors, with 90% of lesions forming in the small intestine, averaging at 2.5 tumors per mouse. Three percent of lesions were localized to the periampullar region, and 7% formed in the colon. Histologically, lesions were characterized as either tubuloglandular adenomas or malignant adenocarcinomas. Adenomas presented with moderate to severe epithelial dysplasia. Adenocarcinomas were associated with prominent stromal reaction and invasion to the muscularis mucosa or to the inner layer of the muscularis. Also, aberrant crypt foci (ACF) were detected in colons. Of interest, seven tumors were tested for mutations in p53 and loss of heterozygosity at the Apc locus. Of these seven tumors, three showed p53 mutations and five were found negative for loss of heterozygosity at the Apc locus; two were uninformative for the markers tested. As this model recapitulates many aspects of human CRC, it provides a good tool for studying the impact of genetic mutations in tumor formation.
Chemically induced models
Chemically induced models of CRC are widely used for studying colitis-associated and sporadic CRC and can involve the use of chemicals such as dextran sulfate sodium (DSS) and azoxymethane (AOM). Models involving these chemicals will be described below.
Dextran sulfate sodium (DSS)
DSS is a non-genotoxic colonic carcinogen, and is commonly used in animal models of colitis. Its exact mode of effect, however, is not presently known, but there is a loss of colonic epithelial cell adhesion 3 days after DSS administration, suggesting that a key effect is the loss-of-barrier function. 27 In addition, a gradual loss of the tight junction protein ZO-1 has been observed over 7 days from the commencement of DSS administration. 28 DSS is also cytotoxic, but it is not known whether this is a result of the changes to cell adhesion and permeability or a direct effect of the DSS. These changes are observed prior to the onset of inflammation, and it is widely believed that the inflammation is thus a secondary effect of DSS. It should be noted, however, that DSS is available in different molecular sizes and its size can affect the inflammatory outcome; DSS of molecular weight of 5 and 40 kD are noted to result in inflammation in contrast to the 500-kD variant. 29
The oral administration of DSS in the drinking water of rodents can induce colitis, and this is the most widely used model for investigating both acute and chronic intestinal inflammation. There are, however, strain differences to the DSS treatment effect, with C57Bl/6 mice being a lot more susceptible to DSS-induced colitis than BALB/c mice. 30 The anatomical sites for colitis also tend to vary between mice strains. For example, inflammation develops in the proximal, middle, and distal colon of C57Bl/6 mice, whereas the cecum, proximal, middle, and distal colon are the primary active sites of inflammation in C3H/HeJBir and NOD-LtJ mice. 31
Chronic intestinal inflammation is associated with the induction of intestinal carcinogenesis. Long-term exposure to DSS alternating with plain water over 204 days was noted to result in the development of flat lesions, which are similar to human colitis-associated CRC, with a 31% incidence of dysplasia and a 25% cancer incidence in the imprinting control region of mice. 32 Unfortunately, the duration to tumor induction in this colitis-associated CRC model is long, with a relatively low incidence and multiplicity of dysplasia and cancer.
Azoxymethane (AOM)
AOM is an organotropic carcinogen and, along with its metabolites, causes DNA adducts via alkylation, forming N 7 -methyl-guanine and O 6 -methyl-guanine, which results in a guanine-to-adenine transition. AOM is injected intraperitoneally into mice, absorbed into the bloodstream, and then transported to the liver. Cytochrome P450 then metabolizes AOM to produce methylazoxymethanol, which is secreted with bile into the gut. It is further metabolized into methyldiazonium by gut microbes, and this is the final carcinogenic metabolite. 33 These activation steps contribute to the organ specificity of AOM and its metabolites, allowing researchers to effectively induce tumors within the intestine.
The concentration of AOM required to induce carcinogenesis is strain-dependent and can vary from 10 to 15 mg/kg, and the colonic tumors then develop over a 24-week period following six once-weekly AOM injections. As it does not rely on the induction of inflammation for tumor growth, this model is thought to mimic the development of sporadic CRC. 33
In AOM-treated rats, ACF, 34 adenomas, and adenocarcinomas develop.34–36 Mutations to the K-ras oncogene are observed 34 along with β-catenin mutations 37 and the absence of full-length APC. 36 Changes in the expression of inducible nitric oxide synthase and cyclooxygenase 2 are also increased in AOM-induced colorectal tumors. 38 These changes are consistent with those found in human CRC, and the AOM-induced model of CRC is widely used in the study of sporadic CRC.
The AOM/DSS model
Modification to the DSS model to include the administration of one of the carcinogens 1,2-dimethylhydrazine or AOM leads to the initial activation of oncogenes. These are genes that promote both cell growth and proliferation. This combination may also result in the inactivation of tumor suppressor genes, which are genes that act to suppress the cell cycle and, therefore, cellular proliferation through the inhibition of growth stimulation factors.
The primary aim of this combined drug model is to provide an intestinal carcinogenesis model where tumors develop over short term and express biological modifications similar to those found in human CRC. This could be achieved with an initial administration of 10 mg/kg AOM, followed by a 1-week exposure to 2% DSS via consumption in the drinking water. 33 This AOM/DSS model mimics the chronic inflammation associated with UC and CD, 39 and results in tumor development, which is dependent on the level of chronic inflammation. Lower levels of DSS concentration in the drinking water, therefore, are associated with decreased numbers of tumors. There are, however, differences in mouse strain sensitivity to AOM/DSS administration, 40 and this model requires further modification for C57BL/6 mice in order to overcome resistance to tumor development. The administration of 10 mg/kg of AOM is thus followed by three cycles of DSS treatment, each cycle consisting of 7 days of 1% DSS in drinking water, followed by plain water for 14 days. This protocol induces visible colonic lesions at day 20 and large tumors in 100% of mice.
Western-style diet-induced models
In addition to genetic mutations and chronic colitis, diet is also a risk factor for CRC development. Diet-based models, therefore, allow the study of specific dietary components on CRC tumorigenesis. One such model is the Western-style diet-induced model of sporadic CRC. 41
In this model, mice were fed a Western-style diet consisting of high dietary lipids and inadequate levels of dietary calcium, vitamin D3, folate, and methyl-donor supplements such as DL-methionine, L-cysteine, and choline bitartrate. Over a 2-year feeding course, approximately 20% of mice developed colonic tumors. Of note is that mice on the Western diet containing adequate levels of dietary calcium and vitamin D3, however, experienced an 89% reduction in overall tumor incidence compared to mice receiving only the Western diet. 42 These findings are supported by a separate study, which modified the Western-style diet (NWD1) to contain elevated levels of calcium and cholecalciferol levels at 7 mg/g and 57 μg/kg (NWD2), respectively. 43 In this study, elevated levels of calcium and cholecalciferol were associated with reduced levels of inflammation when compared to mice on NWD1. However, NWD2 mice also experienced increased weight gain, reduced glucose clearance, altered liver morphology, and a severely altered insulin pathway. In a separate study, changes in inflammation and gene expression were observed in mice fed a Western-style diet consisting of 20% corn oil, 0.5 mg/g calcium, and 0.0125 μg/g cholecalciferol for 3 or 6 months. 44 It was determined that mice on a Western-style diet for 3 and 6 months contained more F4/80-stained macrophages, and gene expression studies on the colon demonstrated an induction of oxidative stress response genes when compared to control mice. Taken together, these results indicate macrophage recruitment and increased oxidative stress as early mechanisms in Western-style diet-induced CRC.
It is of interest that this appears to be the first murine model of sporadic CRC that does not require the presence of a chemical carcinogen or a targeted mutation, suggesting that dietary factors are a risk factor for CRC and that dietary calcium and vitamin D3 might play a role in murine sporadic colorectal tumorigenesis.
Use of Anti-Inflammatory Drugs in Murine Models of CRC
Prostaglandins are inflammatory mediators that are synthesized by cyclooxygenase (COX) isozymes. 45 Prostaglandin synthesis can be inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs), and several studies have utilized NSAIDs in murine models of CRC to determine the effect of inflammation in colorectal tumorigenesis. For example, ApcMin mice treated with piroxicam and sulindac developed 95% and 52% fewer tumors, respectively, when compared to control mice. 46 In this study, it was determined that the main mechanism through which NSAIDs inhibit tumor development was prostaglandin E2 (PGE2), which is synthesized by COX-2. 45 A similar study also reported a dose-dependent reduction in colorectal polyp growth in ApcMin treated with piroxicam. 47 In addition, the formation of colonic polyps in Apcδ716 mice treated with a COX-2-specific inhibitor (3-(3,4-difluoro-phenyl)-4-(4-) methylsulfonyl)phenyl)-2-(5H)-furanone (MF) tricyclic was suppressed in a dosage-dependent manner. 48
It should also be noted that, when comparing the AOM to the AOM/DSS models described in earlier sections, the induction of chronic inflammation through the ingestion of DSS resulted in colonic tumor formation 20 days following treatment, compared to 6 months in the AOM model.
Taken together, these studies demonstrate the pro-tumorigenic role of inflammation. Therefore, studies on the mechanisms of inflammation using the models described here may identify potential therapeutic targets for CRC.
Comparing Animal CRC Models and Human CRC
When developing murine models of IBD-associated and sporadic CRC, it is ideal for the disease model to replicate as many features of the human disease as possible. That, however, is very difficult, and it is important to understand the limitations of models in order to draw reasonable conclusions from any animal model findings. In order to do this, the key features need to be determined, such as 1) the order of disease progression, 2) the general morphology and histology of lesions, 3) the presence of genetic mutations, 4) the presence of ACF, and 5) the presence of chromosomal instability (CIN), microsatellite instability (MSI), and CpG-island methylator phenotype (CIMP). These are described in human IBD-associated and sporadic CRC, and are also present to varying extents in the various murine models mentioned above.
Sporadic CRC is believed to develop from pre-existing colonic adenomas, and the concept of progression from adenoma to carcinoma was strengthened when similarities between the histology of adjacent noninvasive and invasive lesions were noted. 49 By contrast, IBD-associated CRC progression is thought to follow a dysplasia to carcinoma pathway, as it was noted that carcinomas were found in regions of dysplastic tissue without the development of a discrete adenoma. 50
However, the general morphologies of sporadic CRC and CAC tumors are different. For example, most sporadic tumors tend to be exophytic, whereas IBD-associated CRC lesions are mostly flat lesions that develop in the mucosa.51–53 It has also been observed that flat lesions display a relatively high grade of dysplasia. 54
In addition, the association of mutations with tumor-stage-specific genetic abnormalities has led to the development of a model for the clonal evolution of CRC by the acquisition of sequential mutations. 25 For example, loss of tumor suppressor p53 function is strongly associated with dysplasia, and can be detected in dysplastic epithelia and carcinomas in IBD-associated CRC. 55 By contrast, loss of p53 occurs late in the progression of sporadic CRC as observed by gene mutation and allelic loss of p53 in carcinomas but the presence of wild-type alleles in nonmalignant adenomas. 24 In addition, K–ras mutations tend to occur earlier in the IBD-associated CRC progression pathway 24 but are observed late in sporadic adenoma progression.5,25 These mutations are also noted to be more common in sporadic than IBD-associated CRC. 56 In addition, mutations to the APC gene usually occur prior to K-ras mutations and are detected in very early sporadic adenomas.5,57 The reverse, however, is true for IBD-associated CRC. 50
The histological sequence of normal healthy colonic mucosa through to adenocarcinoma also includes the development of ACF into adenomas followed by the progression to carcinoma. 58 ACFs are early colonic abnormalities that are visible on colonoscopic examination by the use of methylene blue staining.53,58,59 Colitis-associated ACFs are distinct from sporadic CRC-ACFs under magnifying endoscopy, with the ACF boundaries of individual crypts obscured, or indistinct, compared to the clear borders found in sporadic ACFs. Most colitis-ACFs are also distorted in shape in contrast to the round, or oval-shaped, sporadic ACFs. 53 Colitis-ACFs are also genetically distinct to the sporadic ACFs with the colitis-ACFs negative for K-ras mutations and p16 expression unlike the sporadic ACFs, 53 suggesting a different developmental pathway.
CIN is detected very early on in the adenoma–carcinoma sequence of sporadic CRC, 60 and in colitis-associated CRC, CIN occurs as an early event as it is detected in nondysplastic tissue.61,62 Similarly, MSI occurs in both CAC and sporadic CRC and can be detected in the dysplastic colitis-associated lesions as well as in sporadic adenomas. 63 Likewise, CIMP is an early event in the development sporadic CRC, occurring in about 50% of sporadic adenomas and carcinomas. 64 These CIMP+ tumors are characterized by high levels of CpG island methylation, features that are not found as frequently in CAC. 65
Taking into consideration some of these key features of both sporadic and colitis-associated CRC, it is of interest to assess these features in murine models of sporadic and colitis-associated CRC. A summary of various features from several sporadic and colitis-associated CRC models and how they compare with the human disease they represent are given in Table 1. The polyps that developed in the AOM/DSS model of colitis-associated CRC were mostly pedunculated and not flat, as observed in humans. 39 ACFs were also not observed in the DSS-only model of colitis-associated CRC. 32 In murine models of sporadic CRC, such as the AOM-only, APCMin, Smad-mutant, and the Western-style diet-induced models, the presence of ACFs and K-ras mutations are not always observed.8,34,41,42,66,67 It is unknown, however, for several of the models whether certain features such as CIN, MSI, and APCs are present.
A summary of the features presented in each model of CRC. Features are summarized according to the characteristic features of CRC, such as the adenoma–carcinoma sequence for sporadic CRC or the dysplasia–carcinoma development in colitis-associated CRC, the incidence of cancer, and the presence of mutations to p53 and K-ras. The presence of ACF, CIN, and MSI are listed as well. Characteristics that are highlighted and in italics are similar to those of human CRC.
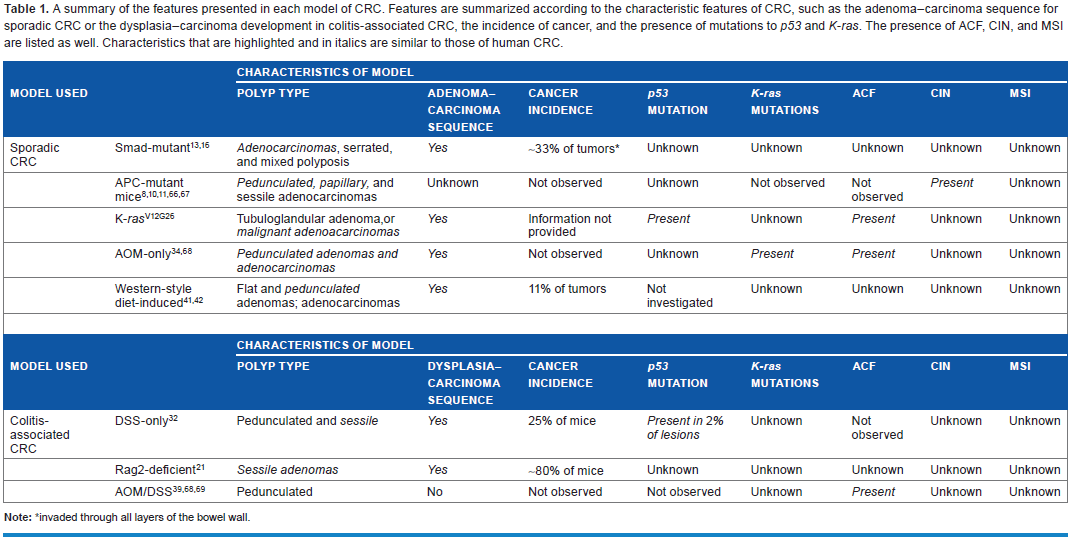
invaded through all layers of the bowel wall.
Conclusion
The models described above have been crucial in understanding sporadic and colitis-associated colorectal tumorigenesis. However, as shown above, there are limitations to each model, which confines research objectives. Therefore, to improve our knowledge of CRC, it is important to fully understand these models and how closely they mimic sporadic and colitis-associated CRC.
Author Contributions
Wrote the first draft of the manuscript: KF. Contributed to the writing of the manuscript: KF, IL. Agree with manuscript results and conclusions: KF, IL. Jointly developed the structure and arguments for the paper: KF, IL. Made critical revisions and approved final version: KF, IL. All authors reviewed and approved of the final manuscript.