
Abstract
Rodent lung tumors are morphologically similar to a subtype of human lung adenocarcinomas. The objective of this study was to evaluate Kirsten rat sarcoma oncogene homolog (Kras), epidermal growth factor receptor (Egfr), and tumor protein 53 (Tp53) mutations, which are relevant to human lung cancer, in cobalt metal dust (CMD)-induced alveolar/bronchiolar tumors of B6C3F1/N mice and F344/NTac rats. Kras mutations were detected in 67% (mice) and 31% (rats) of CMD-induced lung tumors and were predominantly exon 1 codon 12 G to T transversions (80% in mice and 57% in rats). Egfr mutations were detected in 17% (both mice and rats) of CMD-induced lung tumors and were predominantly in exon 20 with 50% G to A transitions (mice and rats). Tp53 mutations were detected in 19% (mice) and 23% (rats) of CMD-induced lung tumors and were predominant in exon 5 (mice, 69% transversions) and exon 6 (rats, all transitions). No mutations were observed for these genes in spontaneous lung tumors or normal lungs from untreated controls. Ames assay indicated that CMD is mutagenic in the absence but not in the presence of S9 mix. Thus, the mutation data (G to T transversions) and Ames assay results suggest that oxidative damage to DNA may be a contributing factor in CMD-induced pulmonary carcinogenesis in rodents.
Keywords
Introduction
Alveolar/bronchiolar adenomas/carcinomas that arise either spontaneously or from chronic chemical exposure are morphologically similar to the adenocarcinoma subtype of the non–small cell lung cancer (NSCLC) in humans (Hong et al. 2008; Pandiri 2015; Tuveson and Jacks 1999). Spontaneous lung (SL) tumor (alveolar/bronchiolar carcinomas) incidence in the B6C3F1/N mouse strain ranges from 9.5% (female) to 27.7% (male; all routes, all vehicles, National Toxicology Program), and the incidence of lung tumors after exposure to certain chemicals was considerably higher in chronic NTP bioassays (NTP 2013a). The incidence of lung tumors in the F344/N rat is considerably lower (1.4% in females and 3.6% in males; all routes, all vehicles) than in the B6C3F1/N mouse (NTP 2013a).
Occupational exposure to cobalt metal is a concern in a variety of metallurgical operations, especially those related to the production of cobalt tungsten carbide and other hard metal alloys. Occupational exposure to cobalt occurs primarily via inhalation of dusts, fumes, and mists containing cobalt. The International Agency for Research on Cancer (IARC) has classified cobalt and cobalt compounds as Group 2B—possibly carcinogenic to humans (IARC 1991). In addition, the epidemiologic data on the role of cobalt in pulmonary carcinogenicity are confounded due to co-exposures in the form of alloys containing tungsten, nickel, and chromium. Although the NTP has studied and confirmed the carcinogenicity of a soluble form of cobalt (cobalt sulfate heptahydrate [NTP 1998]), the contribution of insoluble cobalt metal to pulmonary carcinogenicity is not known. Cobalt metal dust (CMD) was nominated for toxicology and carcinogenesis studies by the United Auto Workers and the Cobalt Development Institute based on the widespread occupational exposure and limited availability of data on chronic toxicity and carcinogenic potential of inhaled insoluble cobalt compounds, particularly CMD. Inhalation was selected as the route of exposure because this is the most common route of exposure to CMD in occupational settings in humans. In the 2-year chronic CMD rodent bioassay, significant dose-related increases in the incidences of lung tumors were observed in both B6C3F1/N mice and F344/NTac rats exposed by inhalation to CMD compared to chamber controls (NTP 2013b). The morphology of alveolar/bronchiolar adenomas/carcinomas that arise spontaneously is indistinguishable from those arising due to chronic CMD exposure.
The process of carcinogenesis involves the alteration of four broad categories of cancer-associated genes: proto-oncogenes, tumor suppressor genes, apoptosis genes, and DNA repair genes (Malarkey, Hoenerhoff, and Maronpot 2013). Chemicals can induce mutations directly by interacting with DNA or indirectly by perturbing basal cellular processes (e.g., by increasing oxidative stress) or by affecting the efficiency of DNA repair. Examining the mutation frequency and spectra of known cancer genes obtained from the tumors of animals exposed to a chemical versus a vehicle control can shed light on the pathway to tumorigenesis engendered by the chemical exposure. However, interpretation of these data is complicated since mutational profiles are dependent on species, strain, gender, tumor differentiation, and dose of carcinogen, as well as dosing regimen.
Lung cancer is a complex disease with variable clinical presentations and behaviors. Genome sequencing of lung cancers from humans has identified several “driver” mutations that may play an important role in lung carcinogenesis. These cancer genes include Kirsten rat sarcoma oncogene homolog in human (KRAS), epidermal growth factor receptor, a protein-coding gene in human (EGFR), tumor Protein 53, tumor suppressor gene in human (TP53), EML4-ALK, HER2 (or ERBB2), BRAF, PIK3CA, PTEN, STK11, FGFR1, DDR2, AKT1, MAP2K1, and MET (Pao and Girard 2011). These genes encode proteins that are critical for cellular proliferation and survival, as well as cellular transformation and tumorigenesis. Of these, KRAS, EGFR, and TP53 are the 3 most commonly altered genes in human lung cancer.
KRAS mutations are observed in about 25% of human NSCLC adenocarcinoma subtype, and of these, the majority of the point mutations are located in codon 12 followed by fewer mutations in codons 13 and 61 (Boch et al. 2013; Rodenhuis et al. 1987). Point mutations in codons 12, 13, and 61 of Kras (Kirsten rat sarcoma oncogene homolog in rodent) are activating mutations that result in constitutive activation of the KRAS protein, making it refractory to the inhibitory guanosine triphosphate hydrolase activating proteins (GAPs). This resistance to inhibition results in stimulus independent, persistent activation of downstream effectors, in particular the Raf-MEK-ERK cascade. Constitutive activation of this kinase cascade results in promotion of cellular proliferation and transformation (Ellis and Clark 2000; Roberts and Der 2007).
The incidence of EGFR mutations in NSCLC adenocarcinoma subtype in humans is about 9% (22/254), with the majority (70%) of the mutations located within exons 19 and 21 (Boch et al. 2013). EGFR is a transmembrane receptor that, upon ligand binding, dimerizes and activates the cytosolic kinase domain of the receptor tyrosine kinase, resulting in activation of signaling pathways that support cancer development and progression. These signaling pathways include the PI3K pathway, which when activated leads to AKT activation and apoptosis inhibition, and the GRB2 and Son of Sevenless (SOS) pathways, which lead to activation of p21RAS, resulting in cell cycle progression (Johnson and Janne 2005).
TP53 is regarded as a master regulator gene that is frequently altered in a wide range of cancers and acts as a tumor suppressor. Its critical roles in cell cycle control, apoptosis, and DNA repair are compromised when the gene is mutated. The incidence of TP53 mutations in human NSCLC adenocarcinoma subtype is ∼50%; the frequency of this mutation is increased in smokers (Husgafvel-Pursiainen and Kannio 1996).
The mutational spectra of TP53 genes obtained from the lung cancers of tobacco smokers exhibit high frequencies of transversions, consistent with the types of miscoding events induced by polycyclic aromatic hydrocarbons present in tobacco smoke, whereas the mutational spectra obtained from the lung cancers of never smokers exhibit transitions (Hernandez-Boussard and Hainaut 1998; Toyooka, Tsuda, and Gazdar 2003). In contrast to KRAS and EGFR mutations, which are predominantly noted mainly in the NSCLC subtype adenocarcinoma, mutations in TP53 are observed equally in both squamous cell carcinoma and adenocarcinoma subtypes of NSCLC (Herbst, Heymach, and Lippman 2008).
Given this knowledge of gene mutations associated with human lung cancers, the objective of the study reported here was to compare mutation frequencies and spectra in alveolar/bronchiolar carcinomas in CMD-exposed rats and mice to the published literature on rodent and human lung cancers. Since the lung tumors from CMD exposure are morphologically indistinguishable from those arising spontaneously, the mutation profiles may provide some information to distinguish the spontaneous lung tumors from those resulting from chemical exposure. Thus, we performed extensive mutation analysis on the Kras, Egfr (epidermal growth factor receptor, a protein-coding gene in rodent), and Tp53 (tumor protein 53, tumor suppressor gene in rodent) genes in lung tumors from B6C3F1/N mice and F344/NTac rats exposed to various doses of CMD by inhalation for 2 years. In addition, traditional bacterial mutagenicity tests, in which positive results are highly correlated with cancer predictivity, were conducted with cobalt metal (the same lot of chemical that was used in the 2-year studies) using three different bacterial tester strains that have different target sites for mutation. Finally, this information may provide mechanistic data to complement the bioassay.
Materials and Methods
Lung Neoplasms
Lung tissue was obtained from an NTP study in which groups of 50 male and 50 female F344/NTac rats and B6C3F1/N mice were exposed to cobalt metal particulate aerosol by inhalation at concentrations of 0, 1.25, 2.5, or 5 mg/m3, 6 hr plus T90 (12 min) per day, 5 days per week for up to 105 weeks (NTP 2013b). Husbandry and experimental procedures were in compliance with the requirements set forth by the Public Health Service’s Guide for the Care and Use of Laboratory Animals. In this study, there was a dose-related increase in the incidence of alveolar/bronchiolar tumors in both male and female rats and mice (Table 1; NTP 2013b). For more details of the animal experiment and pathology data, please refer to the NTP technical report of CMD (NTP 2013b). The criteria used for diagnosing alveolar/bronchiolar carcinomas and adenomas in mice and rats were based on the texts Pathology of the Mouse (Dixon et al. 1999) and Pathology of the Fischer Rat (Boorman and Eustis 1990), respectively. The diagnostic criteria for alveolar/bronchiolar carcinomas outlined in these texts are routinely used by the contract research organizations and are in concordance with the International Harmonization of Nomenclature and Diagnostic Criteria (Renne et al. 2009).
Incidences of alveolar/bronchiolar carcinomas in F344/NTac rats and B6C3F1 mice in the 2-year inhalation study of CMD.
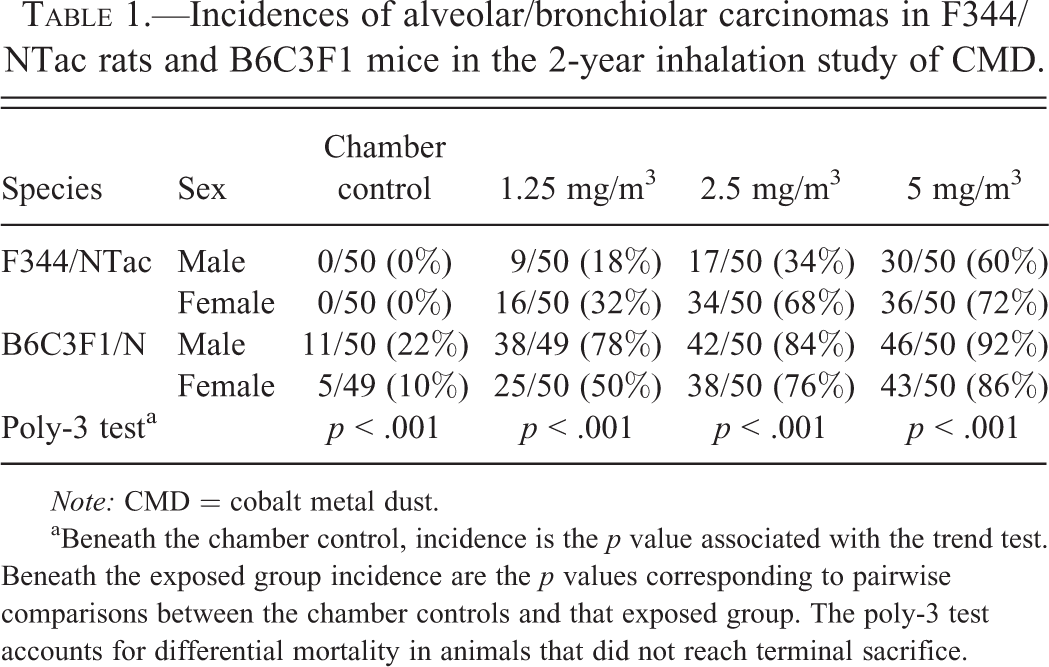
Note: CMD = cobalt metal dust.
aBeneath the chamber control, incidence is the p value associated with the trend test. Beneath the exposed group incidence are the p values corresponding to pairwise comparisons between the chamber controls and that exposed group. The poly-3 test accounts for differential mortality in animals that did not reach terminal sacrifice.
The formalin-fixed paraffin-embedded (FFPE) alveolar/bronchiolar carcinoma tissue was obtained from B6C3F1/N mice and F344/NTac rats exposed to CMD by inhalation for 2 years; spontaneous alveolar/bronchiolar carcinomas from B6C3F1/N mice were obtained from chamber control mice at terminal sacrifice. There were no spontaneous alveolar/bronchiolar carcinomas observed in the chamber control F344/NTac rats in the present cobalt metal study. Since the incidence of SL tumors in F344/N rats is very low (NTP 2013a) compared to the B6C3F1/N mice, the rat tumors (n = 10) had to be sourced from F344/N control groups in various NTP chronic bioassays. As the lung tumor incidence depends on the vehicle in controls (NTP 2013a), we sourced the lung tumors from rats exposed to various vehicles. These studies include sodium azide (animal#VM55), elmiron (animal#VM17), ginseng (animal#VM46), tert-butyl alcohol (animal#VM10; all gavage, water vehicle), probenecid (animal#VF23), isoeugenol (animal#VM3, #VM33; all gavage, corn oil vehicle), trimethylolpropane triacrylate (animal#VM63; topical application in acetone vehicle), lauric acid diethanolamine condensate (animal#VF163; topical application in ethanol vehicle), and ethylbenzene (animal#VM460; inhalation of filtered air). These spontaneous alveolar/bronchiolar carcinoma samples were obtained from 8 male and 2 female F344/N rats. The tumor size determined the sample selection for DNA extraction. If the tumor was greater than 5 mm in diameter, the tumor sample was excised for DNA extraction. On the other hand, if the tumors were smaller (<5 mm in diameter) and randomly scattered throughout the pulmonary parenchyma in a miliary pattern, then the entire section of the lung was used for DNA extraction. Normal lungs without tumors from chamber control B6C3F1/N mice and F344/NTac rats were also collected for histologic evaluation and molecular analysis. After formalin fixation, the tissues were transferred to 70% ethanol and were routinely processed in graded alcohols, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) for microscopic analysis. FFPE tissues were used for mutation analysis. The alveolar/bronchiolar carcinomas were selected for mutation analysis based on their overall size and viability (minimal to no necrosis/hemorrhage microscopically) in order to maximize the amount and quality of DNA obtained from FFPE sections. DNA quality was measured using a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE) to calculate the 260/280 nm absorbance ratio and DNA samples with a purity range of 1.7 to 2.0 were used for analysis. If samples fell outside this range, additional samples were isolated from FFPE sections until a suitable purity measure was obtained or they were discarded.
DNA Extraction, Polymerase Chain Reaction (PCR), Autosequencing, and Mutation Analysis
Alveolar/bronchiolar tumor samples (69 from B6C3F1/N mice and 48 from F344/NTac rats) representing all CMD-exposed dose groups and SL tumors (10 each from B6C3F1/N mice and from F344/N rats) from vehicle control groups were evaluated for hot-spot mutations in Kras, Egfr, and Tp53 genes. FFPE sections at 10-micron thickness were collected on glass slides and the tumors that were greater than 5 mm in diameter were dissected with a sharp microtome blade and collected in screw-top tubes for DNA extraction. Within each lung, if the tumors were multiple and smaller than 5 mm in diameter, then the entire FFPE lung section was collected for DNA extraction. DNA was isolated from these FFPE-dissected tumor tissue sections with DNeasy Tissue Kit (Qiagen, Valencia, CA). Amplification reactions were carried out by semi-nested PCR using primer sets designed for Kras (exons 1 and 2), Tp53 (exons 5–8), and Egfr (exons 18–21; Tables S1 and S2). Controls lacking DNA were run with all sets of reactions. PCR products were purified using a QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). The purified PCR products were cycled with Terminal Ready Reaction Mix-Big Dye (Perkin Elmer, Foster City, CA), and the extension products were purified with DyeEx 2.0 Spin Kit (Qiagen, Valencia, CA). The lyophilized PCR products were sequenced with automatic sequencer (Perkin Elmer ABI Model 3100). The resulting electropherograms were compared to identify mutations in alveolar/bronchiolar carcinomas that arise either spontaneously or due to exposure to CMD. The mutations were confirmed by sequencing with both forward and reverse primers, and the mutations were verified by repeat analysis, starting from amplification of the original DNA extracts.
Bacterial Mutagenicity Studies (Ames Test)
Testing protocols and the principle of the assay have been described in detail in a number of publications (Haworth et al. 1983; Maron and Ames 1983; Zeiger et al. 1992; Mortelmans and Zeiger 2000; Organisation for Economic Cooperation and Development 1997). Positive results in this assay have a high predictivity for carcinogenicity (Tennant et al. 1987), making this test a standard component in all mutagenicity assessments. The assay measures chemical-induced reverse mutation to histidine or tryptophan autotrophy in mutant bacterial tester strains that have lost the ability to autosynthesize these amino acids. Coded samples of cobalt (same chemical used in the NTP 2-year bioassay), dissolved in water, were incubated with histidine-dependent Salmonella typhimurium (TA98, TA100) or tryptophan-dependent Escherichia coli (WP2 uvrA pKM101) tester strains (Moltox, Inc., Boone, NC) either in buffer or S9 mix (metabolic activation enzymes and cofactors from Aroclor 1254-induced male Sprague-Dawley rat liver, Moltox, Inc.) for 20 min at 37°C. Top agar supplemented with
In this assay, data are not subjected to statistical analysis. Rather, a positive response is defined as a reproducible, dose-related increase in revertant colonies in any single strain/activation combination. An equivocal response is defined as an increase in revertants that is not dose related, reproducible, or of sufficient magnitude to support a determination of mutagenicity. When no increase in revertant colonies is observed following chemical treatment, the test is negative. There is no minimum percentage or fold increase required for a chemical to be judged positive.
Statistics
To test for significance of exposure concentration-related trends in the incidences of mutations in lung tumors, a one-sided Cochran–Armitage test was conducted. A Fisher’s exact test was conducted to test for significant differences in the number of mutations between the control and various exposure groups.
Results
Lung Tumors in B6C3F1/N Mice and F344/NTac Rats Chronically Exposed to CMD Harbor Kras, Egfr, and Tp53 Mutations
The incidence of Kras mutations in lung tumors from B6C3F1/N mice chronically exposed to CMD was 67% (46/69; Table 2, Table S3 and S4). The majority of Kras mutations were localized within codon 12 (43%, 30/69), followed by codons 61 (20%, 14/69) and 13 (6%, 4/69). The most common codon 12 mutation in lung tumors from all CMD exposed dose groups was a G to T transversion (GGT to G
Kras mutations in B6C3F1/N mouse alveolar/bronchiolar carcinomas from the chronic CMD bioassay.a
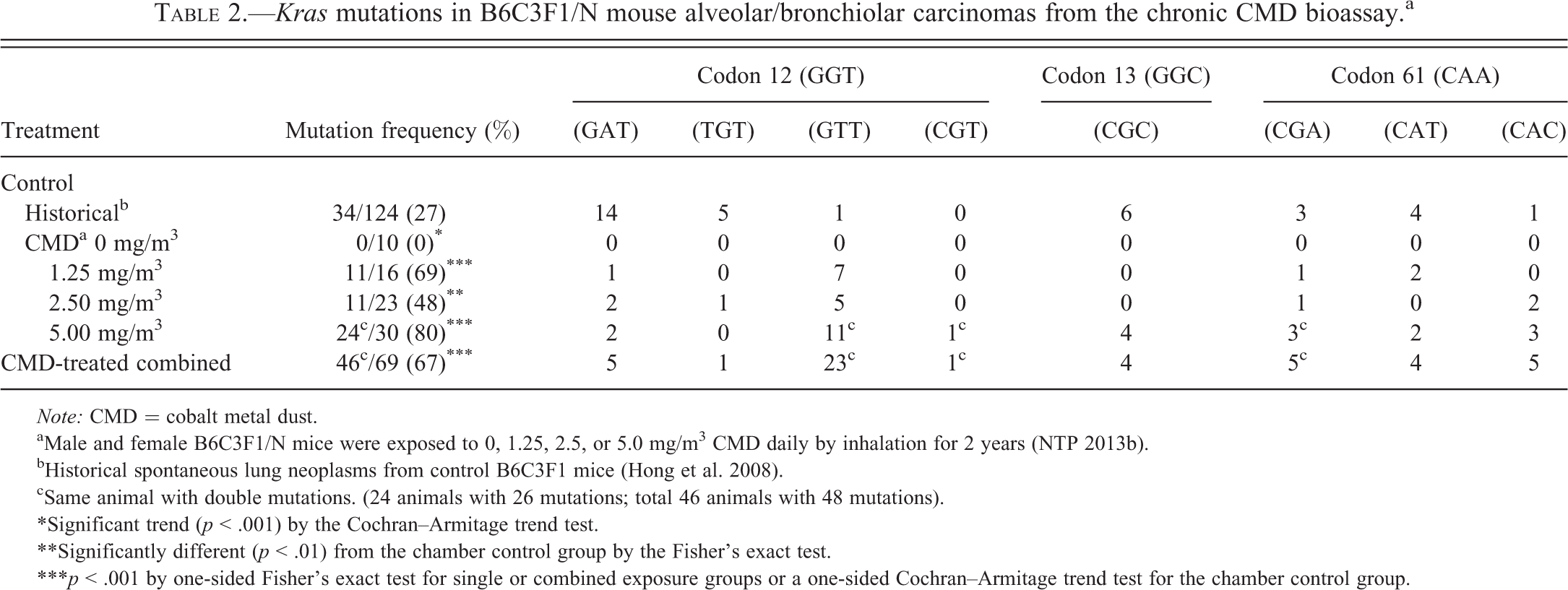
Note: CMD = cobalt metal dust.
aMale and female B6C3F1/N mice were exposed to 0, 1.25, 2.5, or 5.0 mg/m3 CMD daily by inhalation for 2 years (NTP 2013b).
bHistorical spontaneous lung neoplasms from control B6C3F1 mice (Hong et al. 2008).
cSame animal with double mutations. (24 animals with 26 mutations; total 46 animals with 48 mutations).
*Significant trend (p < .001) by the Cochran–Armitage trend test.
**Significantly different (p < .01) from the chamber control group by the Fisher’s exact test.
***p < .001 by one-sided Fisher’s exact test for single or combined exposure groups or a one-sided Cochran–Armitage trend test for the chamber control group.
The incidence of Kras mutations in lung tumors from F344/NTac rats chronically exposed to CMD was 31% (15/48; Table 3, Table S5 and S6). The frequency of Kras mutations was 14% (2/14), 35% (6/17), and 41% (7/17) in lung tumors from 1.25, 2.50, and 5.00 mg/m3, respectively. As in mice, the majority of Kras mutations in the rats were within codon 12 (29%, 14/48), followed by codon 13 (2%, 1/48). Unlike in mice, there were no mutations within codon 61. Similar to mice, the most common codon 12 mutation in rat lung tumors from all CMD-exposed dose groups was a G to T transversion (57%, 8/14), followed by a G to A transition (43%, 6/14). Similar to mice, Kras mutations were not detected in rat SL tumors in this study. There was a dose-related increase in Kras mutations in rats. No Kras, Egfr, and Tp53 mutations were noted in control age-matched F344/NTac normal lung tissues.
Kras mutations in F344/NTac rat alveolar/bronchiolar carcinomas from the chronic CMD bioassay.a

Note: CMD = cobalt metal dust.
aMale and female F344/NTac rats were exposed to 1.25-, 2.5-, or 5.0-mg/m3 CMD daily by inhalation for 2 years (NTP 2013b).
bThere were no spontaneous alveolar/bronchiolar carcinomas in the CMD study. Hence, the spontaneous alveolar/bronchiolar carcinomas (n = 10) were sourced from vehicle or chamber control groups in various NTP chronic bioassays. These spontaneous alveolar/bronchiolar samples were sourced from 8 male and 2 female F344/N rats.
*Significantly different (p < .05) from the chamber control group by the Fisher’s exact test.
**Significant trend (p < .001) by the Cochran–Armitage trend test.
The incidence of Egfr and Tp53 mutations in lung tumors from CMD-exposed B6C3F1/N mice was relatively low. The incidence of Egfr mutations in mouse lung tumors due to chronic CMD exposure was 17% (12/69; Table 4, Table S3 and S4). The frequency of Egfr mutations in mouse lung tumors exposed to CMD was 13% (2/16), 30% (7/23), and 10% (3/30) from 1.25, 2.50, and 5.00 mg/m3 dose groups, respectively. All regions of DNA that were queried for mutations (i.e., exons 18–21) encode the tyrosine kinase domain. The majority of Egfr mutations within lung tumors from CMD-exposed groups were present within exon 20 (9%, 6/69) followed by exon 21 (6%, 4/69), and exons 18 and 19 (each 1%, 1/69). A majority of Egfr mutations in mouse lung tumors were transitions, such as G to A (50%, 6/12) or C to T (17%, 2/12) mutations. Egfr mutations were not detected in mouse SL tumors in this study.
Egfr mutations in B6C3F1/N mouse alveolar/bronchiolar carcinomas from the chronic CMD bioassay.a
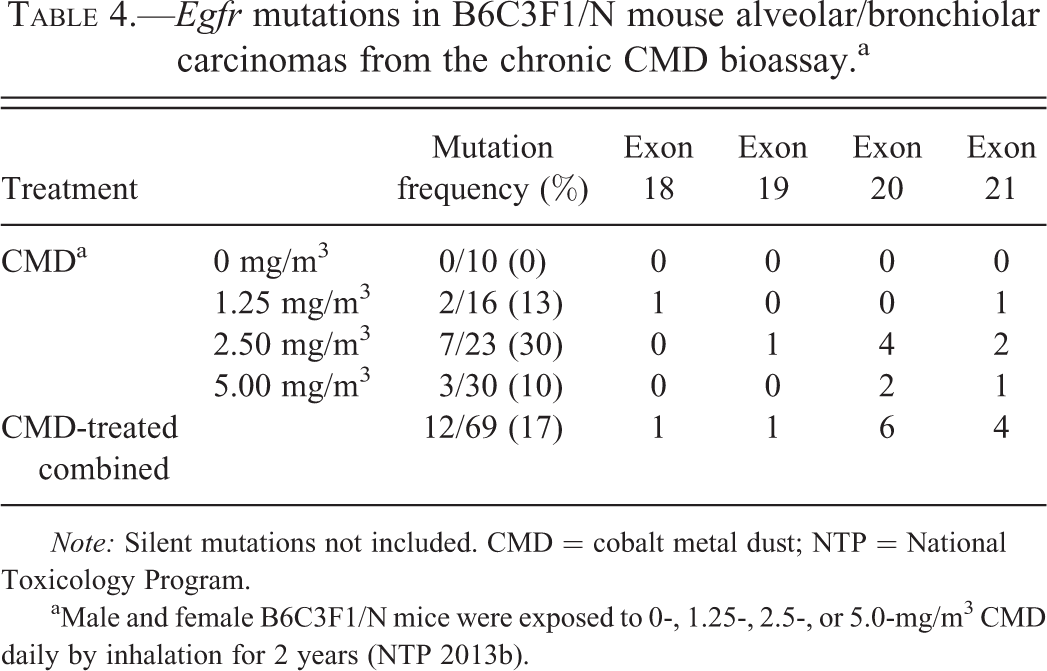
Note: Silent mutations not included. CMD = cobalt metal dust; NTP = National Toxicology Program.
aMale and female B6C3F1/N mice were exposed to 0-, 1.25-, 2.5-, or 5.0-mg/m3 CMD daily by inhalation for 2 years (NTP 2013b).
The incidence of Egfr mutations in lung tumors from F344/NTac rats chronically exposed to CMD was 17% (8/48; Table 5, Table S5 and S6). The frequency of Egfr mutations in rat lung tumors exposed to CMD was 14% (2/14), 18% (3/17), and 18% (3/17) from 1.25, 2.50, and 5.00 mg/m3 dose groups, respectively. As in mice, the majority of Egfr mutations in lung tumors from rats chronically exposed to CMD were present in exon 20 (13%, 6/48), followed by exon 21 (4%, 2/48) and exon 19 (2%, 1/48). A majority of Egfr mutations in rat lung tumors were transitions, such as G to A (50%, 5/10) or C to T (40%, 4/10). Egfr mutations were not detected in rat SL tumors in this study.
Egfr mutations in F344/NTac rat alveolar/bronchiolar carcinomas from the chronic cobalt metal dust (CMD) bioassay.a
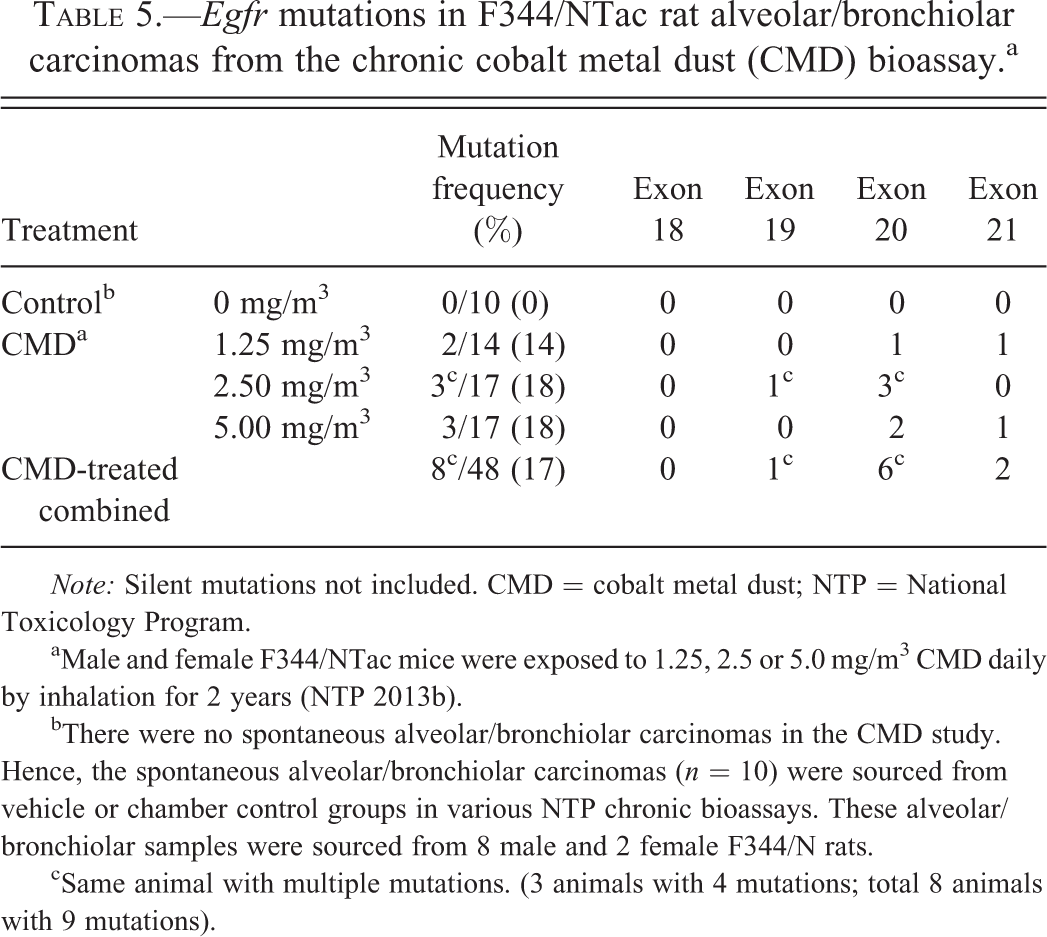
Note: Silent mutations not included. CMD = cobalt metal dust; NTP = National Toxicology Program.
aMale and female F344/NTac mice were exposed to 1.25, 2.5 or 5.0 mg/m3 CMD daily by inhalation for 2 years (NTP 2013b).
bThere were no spontaneous alveolar/bronchiolar carcinomas in the CMD study. Hence, the spontaneous alveolar/bronchiolar carcinomas (n = 10) were sourced from vehicle or chamber control groups in various NTP chronic bioassays. These alveolar/bronchiolar samples were sourced from 8 male and 2 female F344/N rats.
cSame animal with multiple mutations. (3 animals with 4 mutations; total 8 animals with 9 mutations).
The incidence of Tp53 mutations in lung tumors from B6C3F1/N mice chronically exposed to CMD was 19% (13/69; Table 6, Table S7 and S8). The frequency of Tp53 mutations was 19% (3/16), 13% (3/23), and 23% (7/30) in lung tumors from the 1.25, 2.50, and 5.00 mg/m3 dose groups, respectively. The majority of Tp53 mutations in lung tumors from CMD-exposed mice were found within exon 5 (10%, 7/69), followed by exon 7 (6%, 4/69) and exon 6 (4%, 3/69). The majority of mutations were transversions (69%, 9/13), most of which were G to C. Tp53 mutations were not detected in mouse SL tumors in this study.
Tp53 mutations in B6C3F1/N mouse alveolar/bronchiolar carcinomas from the chronic CMD bioassay.a
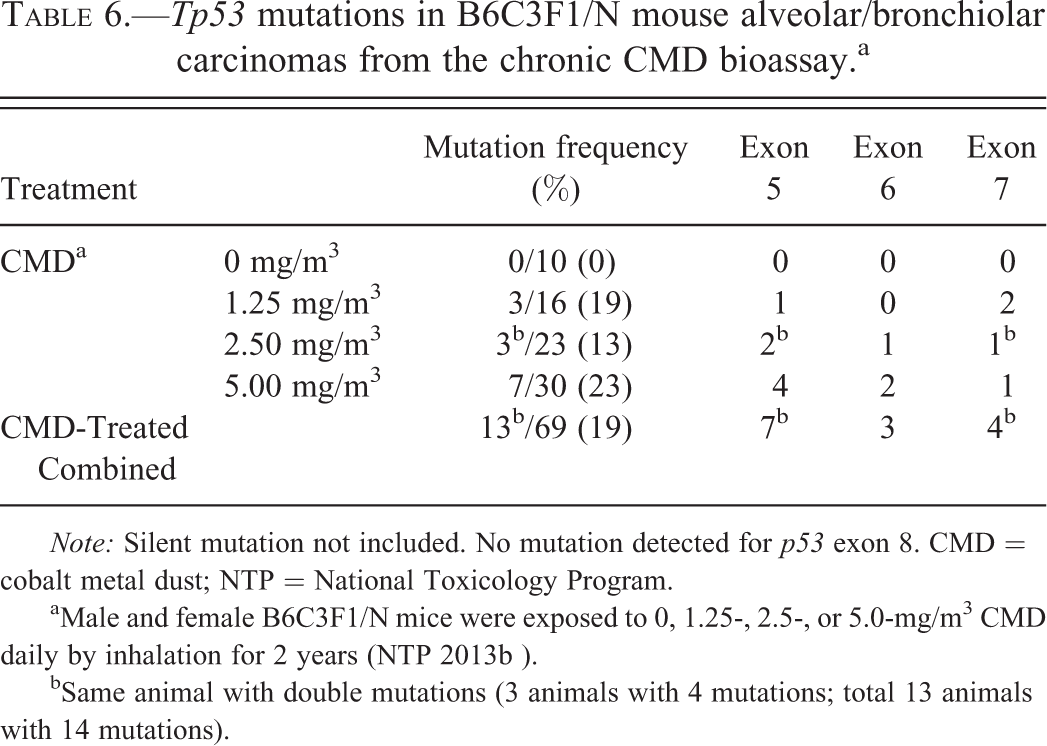
Note: Silent mutation not included. No mutation detected for p53 exon 8. CMD = cobalt metal dust; NTP = National Toxicology Program.
aMale and female B6C3F1/N mice were exposed to 0, 1.25-, 2.5-, or 5.0-mg/m3 CMD daily by inhalation for 2 years (NTP 2013b ).
bSame animal with double mutations (3 animals with 4 mutations; total 13 animals with 14 mutations).
The incidence of Tp53 mutations in lung tumors from CMD-exposed F344/NTac rats was 23% (11/48; Table 7, Table S9 and S10). The frequency of Tp53 mutations was 21% (3/14), 35% (6/17), and 12% (2/17) in the lung tumors from 1.25, 2.50, and 5.00 mg/m3 dose groups, respectively. The majority of Tp53 mutations in rat lung tumors from CMD-exposed groups were present within exon 6 (10%, 5/48), followed by exon 7 (6%, 3/48) and exon 8 (6%, 3/48). In contrast to Tp53 mutations in mice, all Tp53 mutations in rats were transitions, such as C to T (55%, 6/11) or G to A (45%, 5/11). Tp53 mutations were not observed in F344/N rat SL tumors in this study.
Tp53 mutations in F344/NTac rat alveolar/bronchiolar carcinomas from the chronic CMD bioassay.a
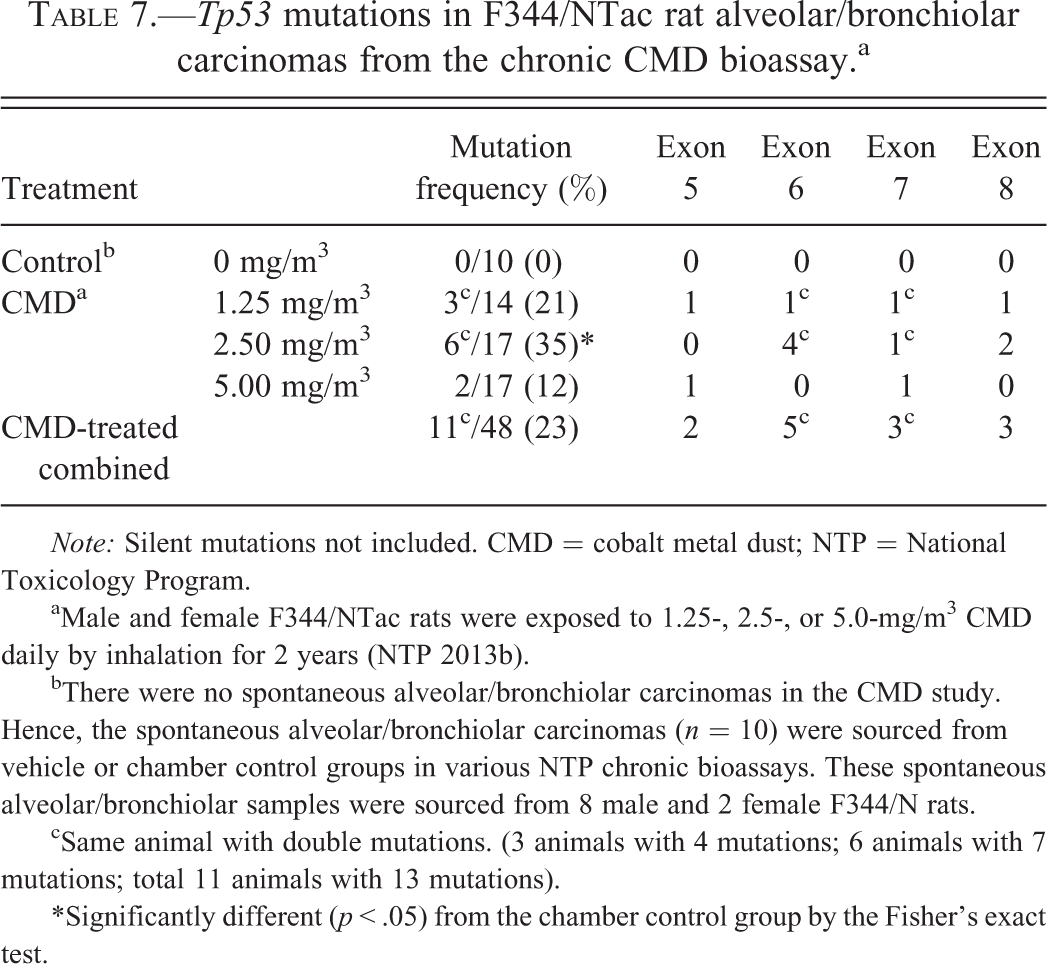
Note: Silent mutations not included. CMD = cobalt metal dust; NTP = National Toxicology Program.
aMale and female F344/NTac rats were exposed to 1.25-, 2.5-, or 5.0-mg/m3 CMD daily by inhalation for 2 years (NTP 2013b).
bThere were no spontaneous alveolar/bronchiolar carcinomas in the CMD study. Hence, the spontaneous alveolar/bronchiolar carcinomas (n = 10) were sourced from vehicle or chamber control groups in various NTP chronic bioassays. These spontaneous alveolar/bronchiolar samples were sourced from 8 male and 2 female F344/N rats.
cSame animal with double mutations. (3 animals with 4 mutations; 6 animals with 7 mutations; total 11 animals with 13 mutations).
*Significantly different (p < .05) from the chamber control group by the Fisher’s exact test.
One male mouse (M605) and one male rat (M408) had multiple mutations (Kras, Egfr, and Tp53), and 3 mice (M219, M605, and F517) and 3 rats (M408, F701, and F723) had Kras and Egfr mutations.
CMD Lung Tumors Share Similar Kras Mutation Spectra with Lung Tumors Resulting from Exposure to Other Chemicals
The Kras mutations in alveolar/bronchiolar carcinomas in B6C3F1/N mice resulting from chronic CMD exposure were compared to the Kras mutations in lung tumors from other NTP studies. The most common Kras mutation in CMD lung tumors was a codon 12 G to T transversion. Similar codon 12 G to T transversions were also noted in cobalt sulfate heptahydrate 75% (6/8), ozone 70% (7/10), ethylene oxide 91% (21/23), and cumene 64% (16/25; Table 8).
Summary of Kras mutations in alveolar/bronchiolar tumors in B6C3F1 mice from various NTP bioassays.
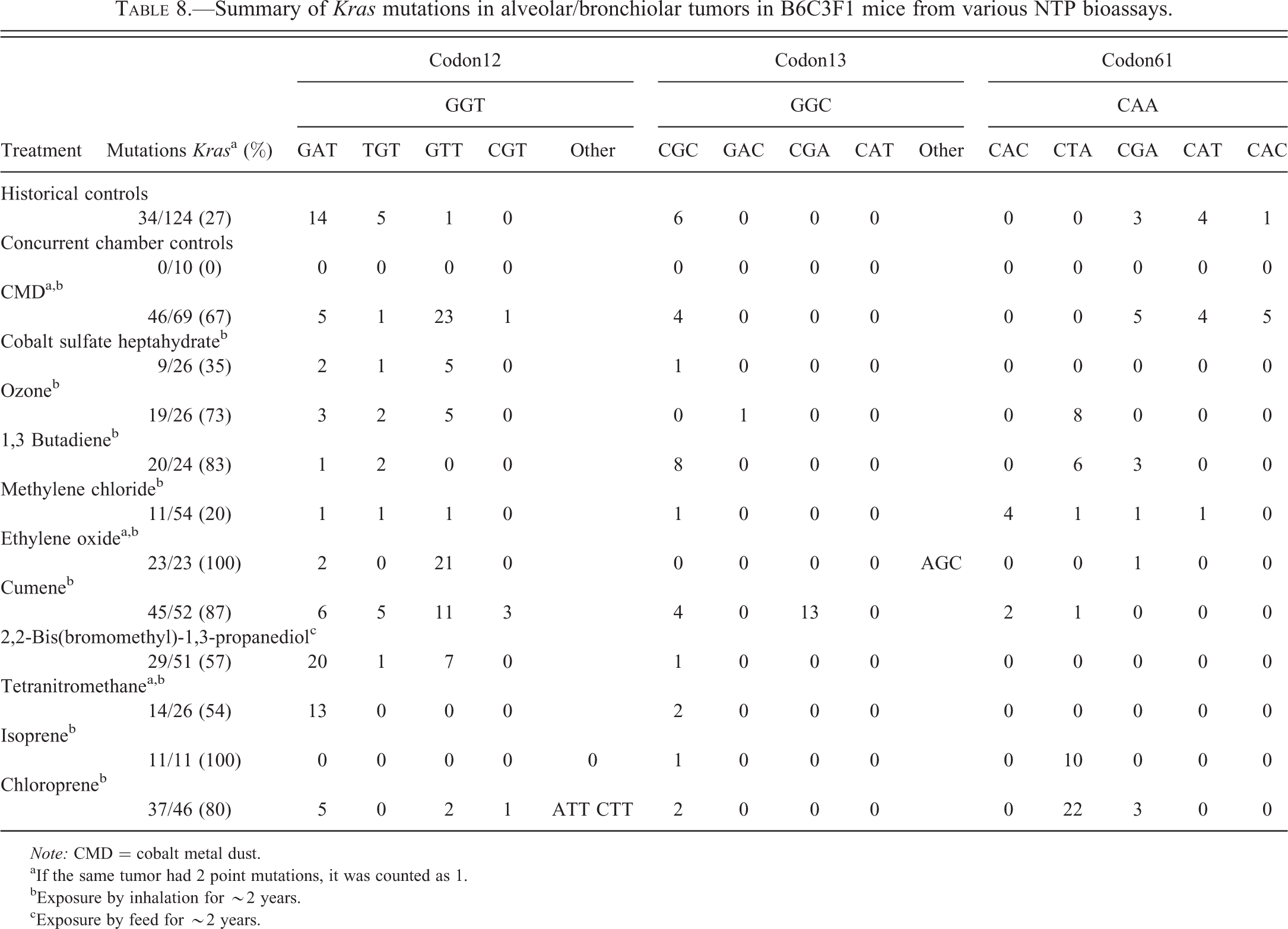
Note: CMD = cobalt metal dust.
aIf the same tumor had 2 point mutations, it was counted as 1.
bExposure by inhalation for ∼2 years.
cExposure by feed for ∼2 years.
CMD Is Mutagenic in the Ames Test in the Absence but Not the Presence of S9 Fraction
In the bacterial gene mutation (Ames) tests conducted with CMD, mutagenic responses were seen in S. typhimurium strain TA100, which mutates via base-pair substitution at a (G:C)3 target, and in strain TA98, which mutates via frameshifting at a dinucleotide run of (CG)4, in the absence of induced rat liver S9 metabolizing enzymes; results for both strains were negative with the addition of S9 mix (Table 9). Negative results were obtained in the E. coli tester strain (WP2 uvrA/pKM101), which mutates via base-pair substitution at the trpE (ochre) TAA codon, in the absence or presence of S9 mix (Table 9).
Mutagenicity of cobalt metal in bacterial tester strains.

Note: NTP = National Toxicology Program; SEM = standard error of the mean.
aMean ± SEM from 3 replicate plates per dose level. Two replicate trials; a representative trial is shown. Complete data set in NTP (2013b).
bTen percent phenobarbital/benzoflavone-induced male Sprague-Dawley rat liver S9 mix for metabolic activation of test article.
cPositive controls were sodium azide (−S9) and benzo[a]pyrene (+S9) for S. typhimurium TA100; 2-nitrofluorene (−S9) and 2-aminoanthracene (+S9) for TA98; and 4-nitroquinoline-N-oxide (−S9) and 2-aminoanthracene (+S9) for E. coli WP2.
dCytotoxicity.
eND = Not Determined.
Discussion
A significantly increased frequency of Kras mutations was observed in CMD-treated B6C3F1/N mice and F344/NTac rats. Kras mutations were more frequent than Tp53 and Egfr mutations within the lung tumors from B6C3F1/N mice and F344/NTac rats chronically exposed to CMD. Surprisingly, no Kras mutations were observed in the control groups in this study. However, Kras mutations in spontaneous mouse lung tumors from historical control data (Hong et al. 2008) were available for comparison to the Kras mutations in lung tumors from CMD exposures. It is interesting to note that in mice, mutations within codon 12 of Kras were observed in both historical SL tumors (27% [34/124]) and lung tumors from CMD-exposed mice (67% [46/69]). However, lung tumors from CMD-exposed mice had predominantly G to T transversions (80% [24/30]), whereas the historical SL tumors had G to A transitions (70% [14/20]) in codon 12. Not surprisingly, G to T transversions were also the most predominant (67% [6/9]) mutations in lung tumors from mice chronically exposed to cobalt sulfate heptahydrate aerosols (Table 8; NTP 1998). Interestingly, G to T transversions are one of the most common Kras mutations in human lung cancer (Rodenhuis et al. 1987). Point mutations at codon 12 of the Kras gene are activating mutations, rendering RAS insensitive to the down-regulatory action of GAPs, thereby locking the protein in a persistently activated state and promoting cellular transformation (Ellis and Clark 2000).
G to T transversions are commonly detected DNA base changes associated with reactive oxygen species (ROS) produced during oxidative damage to DNA (Janssen et al. 1993; Shigenaga and Ames 1991; Tchou et al. 1991). G to T Kras mutations appear to correlate with 8-hydroxydeoxyguanine (8-OH-G) adducts that result from oxidative stress (Shigenaga and Ames 1991; Tchou et al. 1991; Kino and Sugiyama 2005). G to T transversions in codon 12 of Kras were also noted previously in lung tumors in mice exposed to various chemicals, such as tobacco-specific nitrosamines, aflatoxin B1, ethylene oxide, ozone, transplacental azidothymidine, and cumene exposure (Ronai et al. 1993; Sills et al. 1995; Donnelly et al. 1996; Bialkowska et al. 2000; Hong et al. 2008). It is known that some carcinogens such as ethylene oxide, ozone, and cumene induce oxidative stress and DNA adducts resulting in somatic point mutations (Marsden et al. 2009; Van der Zee, Dubbelman, and van Steveninck 1987; Hong et al. 2008). It is not surprising that these particular codon 12 G to T Kras mutations were seen in lung tumors from CMD-exposed mice, and not in SL tumors, since cobalt can induce hypoxia and upregulate HIF-1α signaling, thereby modulating inflammatory responses and inducing oxidative stress (Simonsen, Harbak, and Bennekou 2012). Inflammation that results from the upregulation of inflammatory target genes and proteins may lead to cancer. Oxidative stress stimulates signaling pathways such as PI3K/AKT or mitogen-activated protein kinase (MAPK) to activate NFkB or AP-1. Oxidatively damaged nucleotide bases in the genome are responsible for mutations that can potentially lead to carcinogenesis (Hazra et al. 2003). Thus, the G to T transversions that were observed in mice primarily within codon 12 of Kras may be attributable to oxidative stress resulting from chronic CMD exposure.
In addition, the results of the NTP bacterial mutagenicity assays lend support to the possibility that cobalt metal induces tumorigenesis by increasing oxidative stress, with increased levels of 8-OH-G in DNA as one among several adverse cellular effects. S. typhimurium strain TA98, which showed a response judged to be positive, detects −1 frameshifts that disrupt a dinucleotide run of (CG)4 residues; strain TA100, which showed a response judged to be equivocal, detects reverse mutations at a codon for proline (CCC) in hisG46, and the E. coli WP2 uvrA/pKM101 strain, which showed no increase in mutagenic activity in these tests, detects reverse mutations at the trpE ochre (TAA) codon. Taken together, the degree to which cobalt metal was mutagenic in the 3 bacterial tester strains correlated with the ability of each strain to detect mutational events at G:C base pairs. In support of this observation, sequencing of the supF tRNA mutational reporter gene in bacteria exposed to cobalt chloride showed that almost all mutational events (base substitutions and frameshifts) occurred at G:C base pairs (Ogawa et al. 1999). Cobalt metal-induced mutagenicity was not apparent with addition of S9 mix in any bacterial strain tested by the NTP. Although the composition of rat liver S9 mix has not been fully characterized, it contains microsomal and cytosolic enzymes, and could, therefore, contain antioxidant, radical scavenging enzymes such as glutathione peroxidase, glutathione reductase, glutathione-S-transferase, catalase, and superoxide dismutase. The presence of these enzymes in S9 mix may have ameliorated the mutagenic effects of cobalt. Alternatively (or additionally), the absence of cobalt-induced mutagenic activity in the presence of S9 mix might have been due to binding of cobalt to S9 proteins. We are currently planning experiments to understand the absence of genotoxicity of cobalt in the presence S9 mix.
Cobalt has been shown to affect growth factor receptors including EGFR (Harris and Shi 2003) and the mutation or overexpression of EGFR augments the invasive and metastatic characteristics of cancer in lung (Franklin et al. 2002; Kim, Khuri, and Herbst 2001; Wu et al. 1999). EGFR together with KRAS plays an important role in initiating and maintaining the MAPK signaling cascade and other signaling pathways that are relevant to cancer. KRAS mutations in NSCLC are observed more frequently in smokers, while EGFR mutations in lung cancers are most frequently observed in never-smokers and women (Boch et al. 2013). The incidences of Egfr mutations in lung tumors from CMD exposed mice and rats were lower than the incidences of Kras mutations. In contrast to the Kras mutations, which are predominantly transversions, Egfr mutations in both rat and mouse lung tumors were G to A or C to T transitions. This is the first study in which Egfr mutations were examined and reported in the context of chemically induced rodent lung tumors (Kitahashi et al. 2008). Mutations within Egfr and Kras are considered to be mutually exclusive events in human lung and colon cancers (Kosaka et al. 2004; Shigematsu et al. 2005; Shigematsu and Gazdar 2006; Markman et al. 2010), and treatment modalities have been tailored to the Egfr and Kras mutation status of the tumor (Lievre et al. 2006; Heinemann et al. 2013). However, surprisingly, in lung tumors in the CMD-exposed rodents in our study, 25% (3/12) of mice and 38% (3/8) of rats that harbored Egfr mutations also had Kras mutations. Furthermore, one mouse (M605) in the high dose group and one rat (M408) in the medium dose group had multiple mutations (Kras, Egfr and Tp53) in the same animal.
Mutations in Tp53 in chemically induced rodent models are considered a late event, especially in tumors that were initiated by mutations in Kras (Horio et al. 1996). Cobalt may affect Tp53 transcription and expression through DNA damage, DNA binding, the inhibition of DNA repair, gene silencing, and through ROS (Harris and Shi 2003; Zhao et al. 2013; ROC 2011). ROS have been observed to affect signal transductions, including MAPK signaling, and the activation of transcription factors including NFkB, AP-1, and HIF-1 (Harris and Shi 2003). The incidence of Tp53 mutations resulting from chronic CMD exposure was similar in both mice and rats, but the nature of mutations was different, that is, Tp53 mutations in mice were predominantly transversions, whereas in the rat they were predominantly transitions. Interestingly, in humans, TP53 transversions are commonly observed in NSCLC from smokers, while TP53 transitions are often noted in NSCLC from never smokers (Gao et al. 1997; Le Calvez et al. 2005). Tp53 G:C to T:A transversions are the most frequent substitutions and more biologically relevant in human lung cancers (Hollstein et al. 1991). The predominance of CMD-induced alveolar/bronchiolar tumors with Tp53 mutations may provide a selective advantage for unregulated growth and the avoidance of apoptosis (Rodin and Rodin 2005). The significance of this differential mutation spectrum in mice and rats and its relationship to human lung cancer needs to be further explored. Complementary studies (such as performing immunohistochemistry for downstsream markers supporting the alterations within the respective molecular pathways) and further microarray analysis underscoring the complexity of lung cancer should be considered.
In summary, there was a significantly elevated incidence of Kras mutations, accompanied by a lower incidence of Egfr and Tp53 mutations, in lung tumors from mice and rats chronically exposed to CMD compared to SL tumors. The mutations detected in the Kras, Egfr, and Tp53 genes in CMD-exposed mice and rats clearly imply those genetic events are related to chemical exposure, since those mutations were not detected in the concurrent spontaneous tumors in mice and SL tumors from previous NTP studies in rats. Several of the observed mutations in Kras are comparable to mutations observed in other NTP studies (Table 8) in which a significant increase in the incidence of lung tumors in response to chronic chemical exposure was evident. In addition, these mutations arose within the hotspot regions of Kras, Egfr, and Tp53 genes and are thus comparable to the mutations observed in human NSCLC. The significance of these findings is that the genetic alterations in the rodent lung tumors represent similar alterations in cancer genes that play a role in lung cancer in humans. In addition, the positive Ames assay results only in the absence of S9 mix along with a predominant G to T transversions in the lung tumors resulting from CMD exposure suggests that oxidative stress might play a contributing role in pulmonary carcinogenesis of CMD.
Footnotes
Acknowledgments
We also would like to thank Drs. Gregory Travlos and Kyathanahalli Janardhan for internal review of this article.
Author Contribution
Authors contributed to conception or design (MaJH, RH, MiJH, MB, RS, AP); data acquisition, analysis, or interpretation (HH, TT, RH, GK, MB, KW, SS, AP); drafting the manuscript (HH, AP); and critically revising the manuscript (MaJH, MiJH, KW, SS, RS, AP). All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Divisions of the National Toxicology Program and the Intramural Research Program at the NIEHS, NIH. We would like to thank the NIEHS DNA Sequencing Core for their technical expertise and the NTP tissue archives at the Experimental Pathology Laboratories, Inc.
Supplemental Material
Supplemental Table S1. Primers used in amplifying the hot spot regions of mouse Kras, Tp53, and Egfr genes.
Supplemental Table S2. Primers used in amplifying the hot spot regions of rat Kras, Tp53, and Egfr genes.
Supplemental Table S3. Kras and Egfr Mutations in male B6C3F1/N mouse lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S4. Kras and Egfr Mutations in female B6C3F1/N mouse lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S5. Kras and Egfr mutations in male F344/NTac rat lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S6. Kras and Egfr mutations in female F344/NTac rat lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S7. Tp53 Mutations in male B6C3F1/N mouse lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S8. Tp53 Mutations in female B6C3F1/N mouse lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S9. Tp53 mutations in male F344/NTac rat lung from the chronic cobalt metal dust (CMD) bioassay.
Supplemental Table S10. Tp53 mutations in female F344/NTac rat lung from the chronic cobalt metal dust (CMD) bioassay.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.