
Abstract
Colon cancer is a major human malignancy that afflicts millions of people throughout the world each year. Genetics and diet play large roles in colon carcinogenesis although chemicals may also contribute. For the past 40 years, scientists have studied experimentally induced intestinal carcinogenesis in rodents in order to elucidate the etiology and mechanisms involved. Comparative histopathology has revealed many similarities of rodent and human intestinal cancers. Comparative molecular pathology has also shown genetic similarities. More recently, genetically engineered mice and inflammatory colon cancer models have been used for investigating mechanisms and potential chemopreventive and treatment modalities. This review will focus on comparative histopathology and nonclinical models.
Keywords
Introduction
Colon cancer is a major human cancer and cause of death in many countries, especially those with specific types of diets. It is thought that diet, genetics, and other factors play a role in human colon cancer (Derry et al. 2013). Cancer of the small intestine is much less common. The search for the etiology and mechanisms involved in human colon cancer has led to the development of many experimental models in laboratory animals. These experimental models include those induced by chemicals, genetic engineering, and inflammatory disorders. Validation of these animal models by pathologists entails establishing the diagnostic criteria and classifications of the intestinal tumors with comparison to the human condition. Nomenclature of gastrointestinal (GI) tract lesions has been published by various authors and international pathology committees (Boivin et al. 2003; Washington et al. 2013; Whiteley et al. 1996), and the most recent iteration is presently in progress by an INHAND GI Tract nomenclature committee (Nolte et al. 2014). Intestinal lesion definition and nomenclature plays a role in defining preclinical models for colon cancer and inflammatory bowel disease (IBD). This article will review the latest information on these important issues.
Etiology of Intestinal Tumors in Rodents
The causes of intestinal tumors in rodents are well known. Such tumors occur in low incidences in control animals (Chandra, Nolan, and Malarkey 2010) but can be induced by a variety of experimental methods. Several chemicals (more than 30 in rats and more than 9 in mice) cause tumors of the small and large intestines in rodents including those derived from natural product cycasin (azoxymethane [AOM] and dimethylhydrazine), several nitroso compounds, Aloe vera extracts, heterocyclic amines, folpet, captan, dextran sulfate sodium (DSS), and others (Boudreau et al. 2013; Cohen et al. 2010; Gold et al. 2001; Chandra, Nolan, and Malarkey 2010; Pandiri et al. 2011; Ward 1975; Ward, Yamamoto and Brown 1973). Although most of these intestinal carcinogens have low human exposure, Aloe vera solutions for oral use are sold in health food stores.
The general molecular pathogenesis of colon cancer in humans has been reviewed in many publications (Grady 2014; Bosman et al. 2010). Common molecular pathways for some rodent and human colon tumors have been reported (Pandiri et al. 2011; Sills et al. 2004). The role of specific genes in each stage of human colon carcinogenesis, from dysplasia to adenoma to adenocarcinoma (Bosman et al. 2010), has also been suggested by the creation of genetically engineered mice (GEM) and in chemical-induced tumors of rats and mice. Important genes in the human colon carcinogenesis process include APC, CTNNB1, IGF12, TGFBR2, KRAS, BRAF, and TRP53 (Figure 1). Likewise, many lines of GEM develop intestinal tumors including those involving inactivation or modulation of Apc, Smad3, Smad4, Ctnnb1, Pi3k, and other genes (Figure 1), which are also involved in human colon cancer molecular histopathogenesis (Boivin et al. 2003; Deming, Leystra, Farhoud, et al. 2013; Halberg et al. 2009; Hermiston and Gordon 1995; Hung et al. 2010; Kosa et al. 2012; Leystra et al. 2012; Salcedo et al. 2010; Taketo and Edelmann 2009; Washington et al. 2013; Zeineldin and Neufeld 2013; Xue et al. 2010). Many of these cancer mouse lines are available from various sources (http://mouse.ncifcrf.gov; http://jaxmice.jax.org/cancer/index.html).
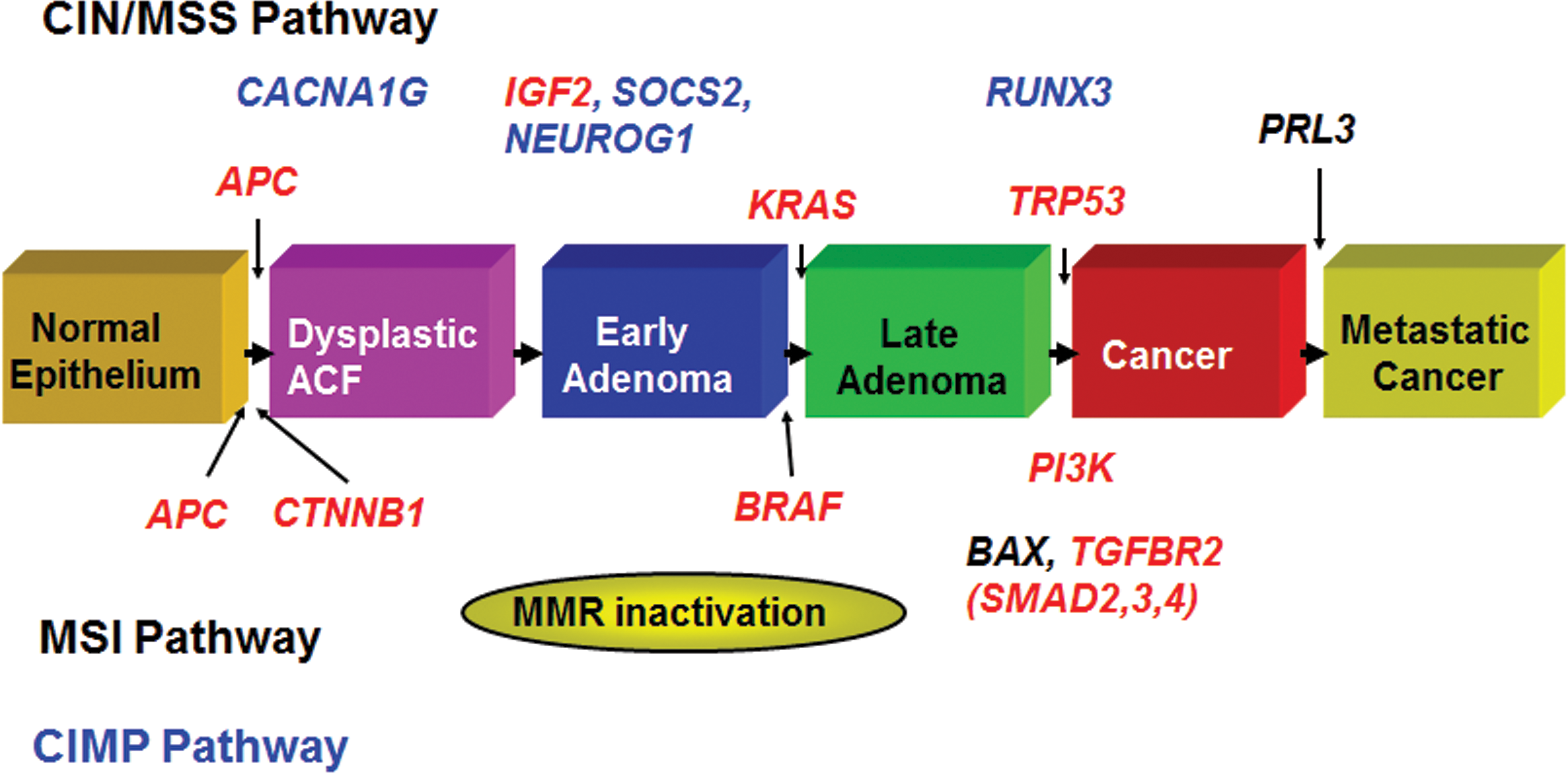
The molecular histopathogenesis of colon cancer in humans, mice, and rats. (modified from and courtesy of William M. Grady and from references noted in the text).
Newer inflammation-associated colon cancer models have been developed especially involving immunodeficient mice and Helicobacter sp. infections (Erdman and Poutahidis 2010; Erdman, Poutahidis, et al. 2003; Maggio-Price et al. 2005, 2006, 2009) and colon irritants such as DSS (Tanaka 2012a, 2012b). DSS alone can cause colonic adenocarcinoma (Cooper et al. 2000). Two lines of rats with a genetically engineered Apc gene have also been created (Amos-Landgraf et al. 2007; Zeineldin and Neufeld 2013). Chemically induced colon tumors in rats exposed to Aloe vera extracts or o-Nitrotoluene were found to have Kras or Ctnnb1 mutations (Sills et al. 2004; Pandiri et al. 2011).
Pathology of Intestinal Tumors
Necropsy, Fixation, and Trimming Protocols
General toxicologic pathology necropsy protocols (Ruehl-Fehlert et al. 2003, http://reni.item.fraunhofer.de/reni/trimming) are usually sufficient to detect intestinal carcinogens in 2-year carcinogenesis studies (Boudreau et al. 2013).
For studies in which intestinal tumors are expected, the entire GI tract may be opened from stomach to anus in order to identify all grossly visible lesions. The GI tract may be flattened onto paper, and then lesion counts and measurements may be performed. Tumors often have the appearance of benign lesions (polyps), which grow into the lumen with stalks (Figures 2 and 3) or grow into the gut wall (Figures 4, 5, and 6), and some may metastasize to the peritoneal cavity and other tissues (Figure 5). The inflammatory colon cancer models often induce mucinous cystic adenocarcinomas (Figure 6). In some chemically induced tumor models of rats and mice, there is diffuse thickening of the entire colon and focal tumors (Figure 7). The diffuse thickening can be due to diffuse epithelial hyperplasia and inflammation and/or early focal neoplastic lesions. After fixation, the gut can be divided into sections and long sections prepared to survey much of the intestine for preneoplastic lesions and tumors. Also, each tumor with adjacent normal tissue for fixation can be identified and placed in a separate small container with fixative and after fixation, trimmed in half. Both halves can be embedded in the same paraffin block and placed on the same slide. The entire tumor is now in the block and multiple sections can be used for various stains. Alternatively, small intestinal and large intestinal gut rolls (Swiss rolls) can be prepared in order to review the entire intestine in 1 to 2 slides (Figure 8). The section of the gut roll has the advantage of being able to see many lesions of various sizes on 1 slide but with the disadvantage of numerous artifacts and tangential sections and often lack cross sections through large tumors. Step and serial sections can be performed to better illustrate tumor morphology and invasion, if any. With questionable invasive lesions, step sections are critical.
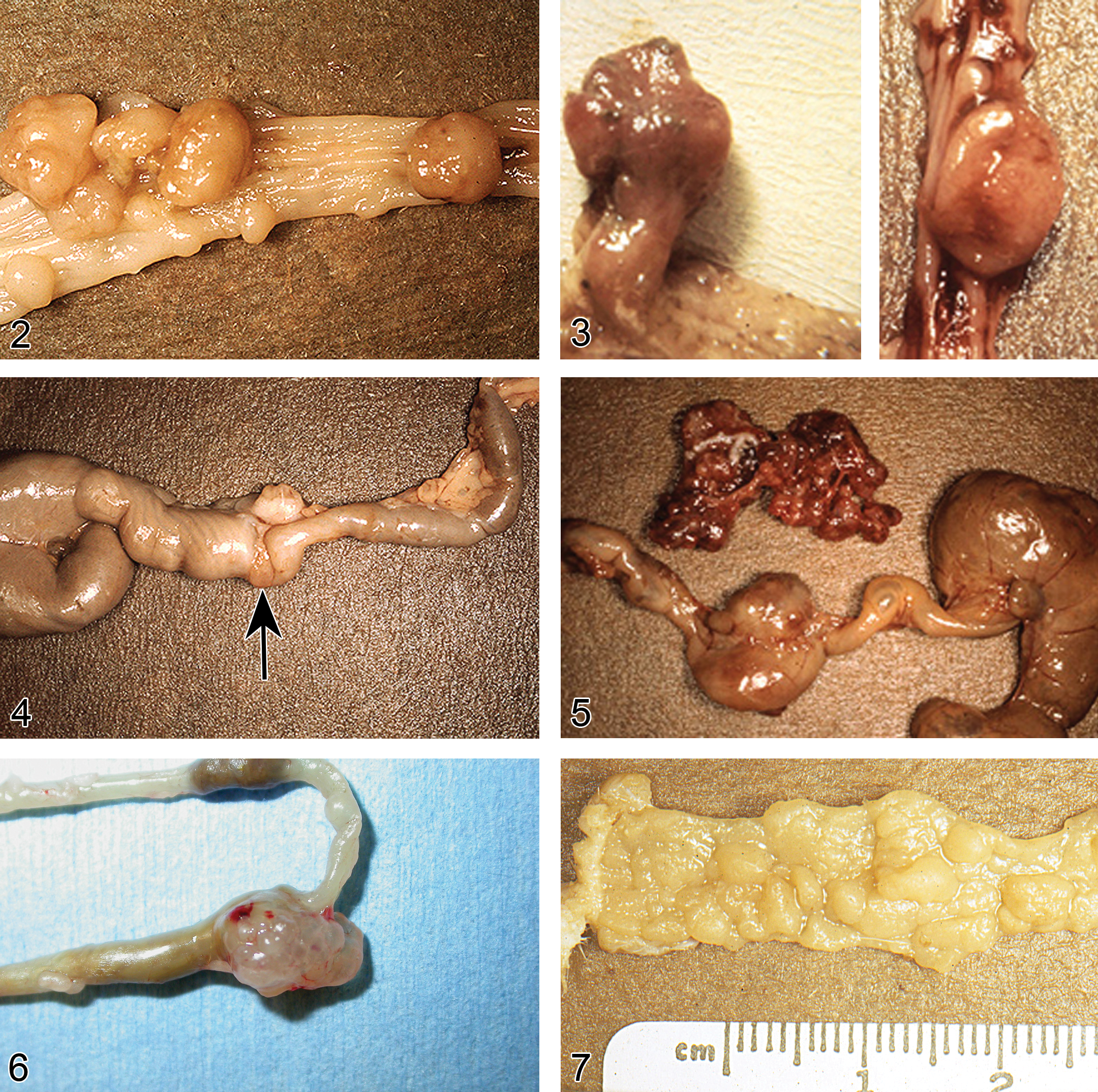
Formalin fixation is the fixative of choice in routine bioassays but for special studies of intestine, Bouin’s fixative allows for optimal sectioning for histopathology but may limit some immunohistochemistry studies. Some of our figures are from Bouin’s-fixed intestinal lesions. Bouin’s-fixed sections are amendable to thin sectioning and also show more intense H&E staining. Necropsy techniques, fixation, and trimming should be tailored to adhere to study goals and requirements.
Tumor Pathology
The pathology of intestinal tumors in rodents is very similar to that in humans (Bosman et al. 2010) and other species (Cardiff et al. 2004, 2006; Ward 1974; Ward and Ohshima 1985), although human colon adenocarcinomas probably metastasize more frequently than those in rodents, in general. Preneoplastic and precancerous lesions in small or large intestine develop through a sequential histopathogenesis and molecular pathogenesis in humans and in various rodent models (chemical, GEM, and inflammatory). Preneoplastic lesions are those changes that may progress by growing in size to noninvasive benign or from benign to invasive lesions. In the rodent colon, noninvasive lesions are usually adenomas of various types. Precancerous lesions are those that may progress in size and behavior to develop into invasive malignant lesions. In humans, there are many precancerous lesions in various tissues but less preneoplastic lesions generally than in rodents. In the human large intestine, a preneoplastic lesion is a microscopic colonic dysplasia that is also common in human familial polyposis cases. In mice, similar lesions are common in Apc mutant mice where they may be clonal or polyclonal (Thliveris et al. 2005).
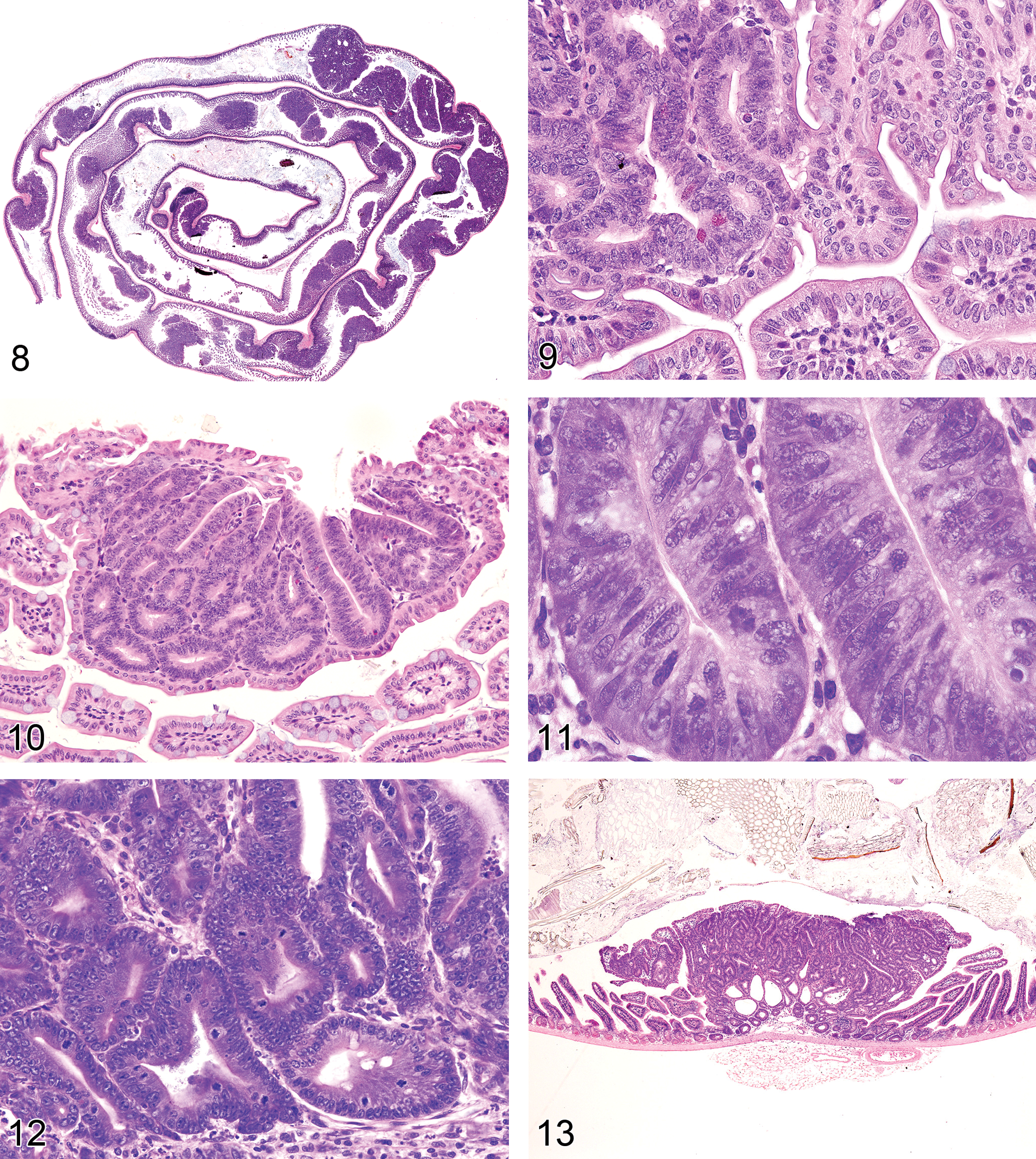
Preneoplastic and precancerous intestinal lesions in rats and mice begin as tiny microscopic foci, not usually visible grossly at the time of necropsy (Figures 9 and 10). These lesions have been most studied in the large bowel of rats and mice but have similar morphological characteristics in both tissues. They have been diagnosed as subgrossly visible aberrant crypt foci only with the use of methylene blue staining and a dissecting microscope and histologically as low- or high-grade dysplasias, atypia, atypical hyperplasia, carcinoma in situ, intraepithelial neoplasia (IEN), and microadenoma (Boivin et al. 2003). IEN is currently out of favor for human colon nomenclature (Washington et al. 2013). For toxicologic pathology nomenclature, “hyperplasia, atypical” is the preferred term, in part, to simplify and consolidate the nomenclature (Betton et al. 2001; Deschl et al. 1997; Nolte et al. 2014; Whiteley et al. 1996; Figures 9 and 10). Mouse intestinal tumor nomenclature using human nomenclature when possible has also been published (Boivin et al. 2003; Boivin and Groden 2004; Washington et al. 2013). The finding of atypical hyperplasias in 2-year rodent carcinogenesis studies is not common. In fact, a recent National Toxicology Program study of Aloe vera extracts revealed an over 60% incidence of colon adenomas or carcinomas in high-dose male rats but not atypical hyperplasias or any lesions diagnosed as preneoplastic (Boudreau et al. 2013, http://ntp.niehs.nih.gov/Ntp/About_Ntp/Trpanel/2011/April/DraftTR577.Pdf) although goblet cell hyperplasia, a nonpreneoplastic lesion, was seen in rats also. Hyperplasia, atypical should be used for toxicologic pathology nomenclature, when possible, for an established preneoplastic lesion in rodents (Figures 9 and 10) and not for regenerative lesions that are often dysplastic in mice and rats, after acute or chronic intestinal injury (Figure 11). Rodent preneoplastic lesions (hyperplasia, atypical) often have the same morphology as surface epithelium of adenomas (Figure 12).
Adenomas may be precancerous, especially in human colon, but are much less so in most rat and mouse intestinal carcinogenesis models. Rodent adenomas may be sessile (Figure 13), polypoid (Figures 14 and 15), or papillary. Adenomas often develop stalks that show evidence of possible invasion of the stalk (Figures 16 and 17). It may appear that the normal structure of the intestinal layers (lamina propria and muscularis mucosa) is disrupted and invaded by tumor cells but often this is not true invasion but only physical disruption, as the tumor grows into the gut lumen (perhaps as a form of herniation). These adenomatous lesions in most rodent models do not progress to invasive adenocarcinomas. The sequential changes in intestinal carcinogenesis are best studied by serial sacrifice of rodents. In most rodent intestinal cancer models, intestinal adenocarcinomas often develop de novo from flat lesions (Figures 18 and 19) and not from adenomas, in an epithelium that has no diffuse inflammatory lesions (colitis), with the exception for the inflammatory colon cancer mouse models, discussed later. A stromal and inflammatory response, however, is often seen during early and late adenocarcinoma development in response to invasive carcinoma cells. These flat early adenocarcinoma lesions are much less common in human colon where adenocarcinoma most often develops from dysplasia in adenomas. As they invade more deeply into the intestinal wall, they show typical behavior and morphology of adenocarcinoma (Figures 20–25).
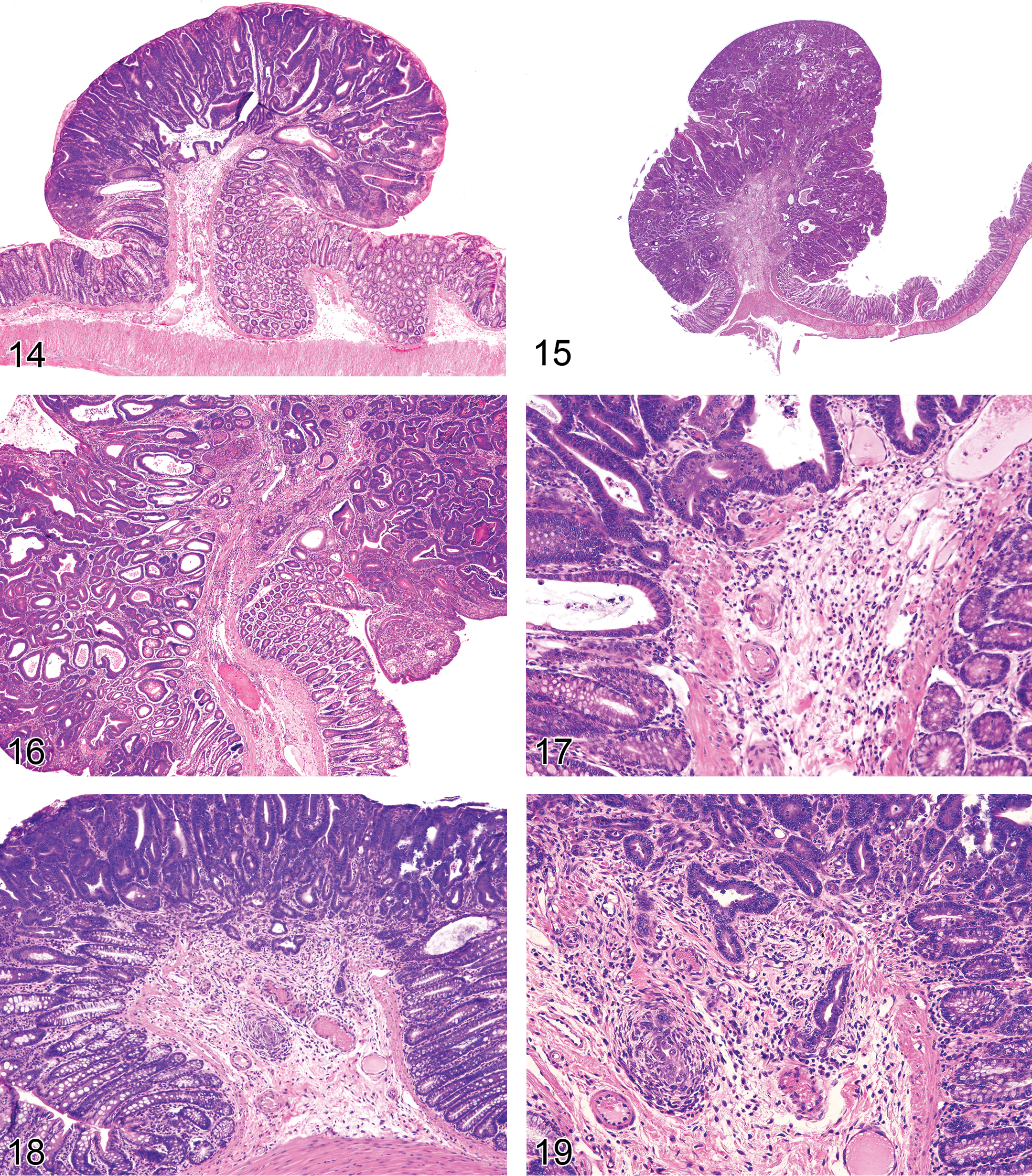
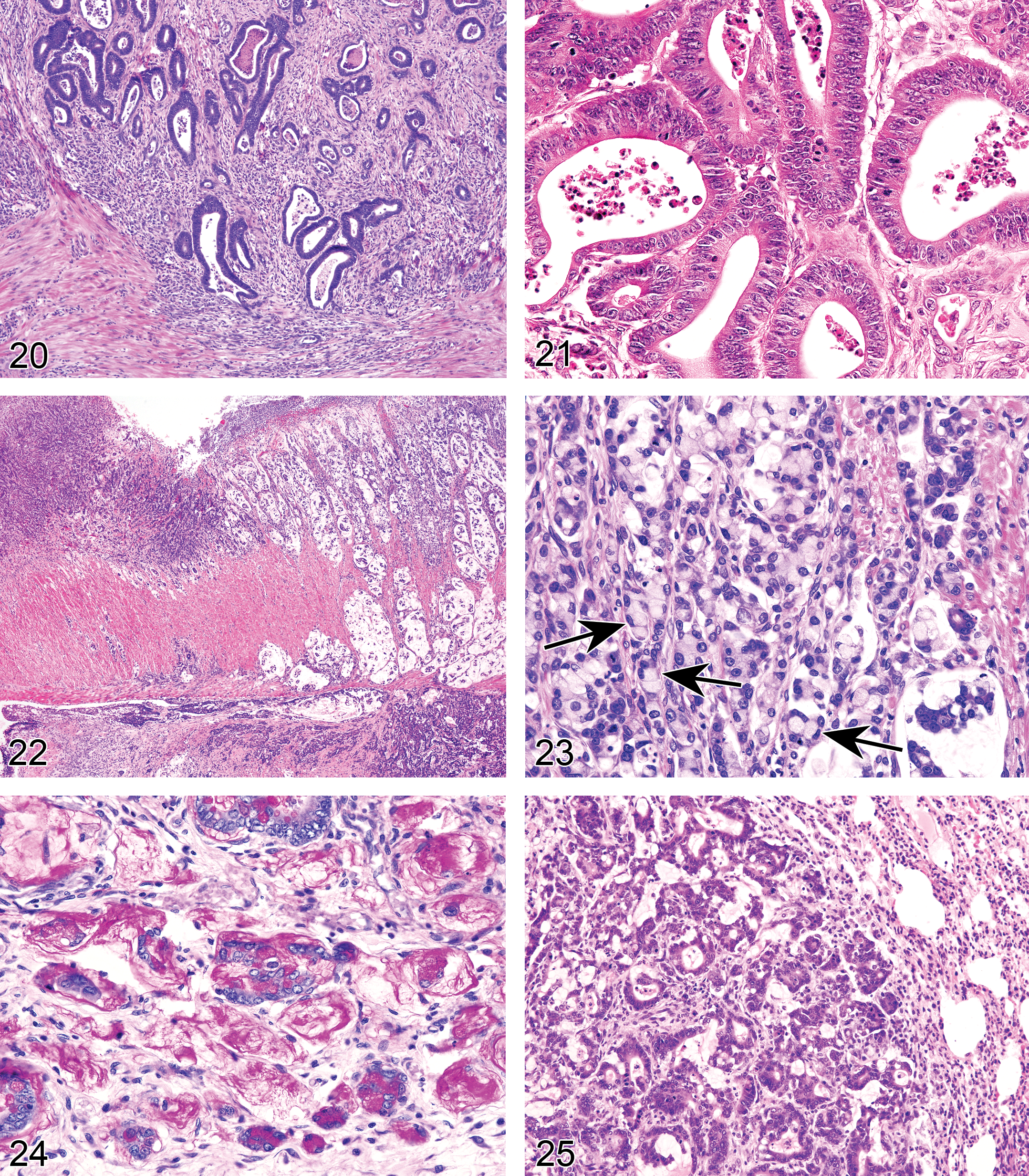
Differentiation of preneoplastic and precancerous intraepithelial microscopic foci from regenerative dysplastic lesions in a colon with inflammatory lesions is challenging. DSS, a colon irritant, causes ulcers after a few days of exposure with marked inflammation and epithelial regeneration adjacent to the ulcers (Figure 11). The acute and chronic regenerative lesions are very dysplastic and resemble preneoplastic foci induced by carcinogens and induced genetic modifications. In more chronic studies with inflammatory models, it may be possible to differentiate these lesions especially when they begin to invade the submucosa and tunica muscularis and serosa (Figures 26–31). Important criteria for malignancy in mouse models are the context of the lesion (focal nature and invasive characteristics), the local environment (angiogenesis, inflammatory response different than the diffuse colitis), and its biological process (have similar lesions been shown to progress?) with lesser emphasis placed only on the morphology of the epithelial cells.
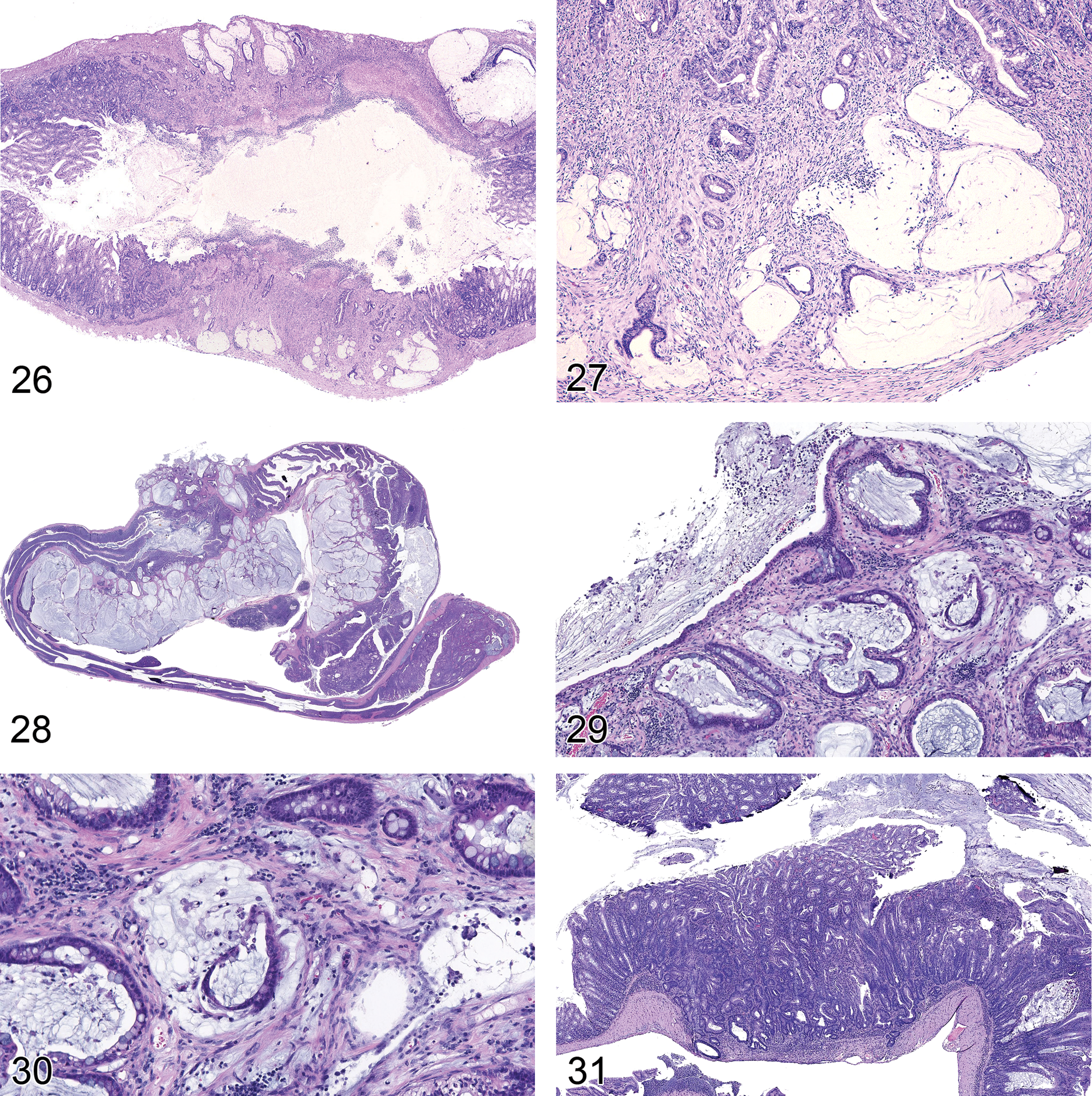
Intestinal adenomas and adenocarcinomas induced by specific chemicals and in GEM often have unique histopathologic types and biological behavior. These findings can be matched by the molecular changes in genes involved in the carcinogenic process. Genotype can induce morphologic phenotype of tumors in general especially for GEM (Cardiff et al. 2004) as it may in human colon tumors (Bosman et al. 2010). Rodent adenocarcinomas may be scirrhous (Figure 20), tubular (Figure 21), papillary, tubular–papillary, mucinous (Figures 22, 23, and 24), signet ring, solid, undifferentiated, or mixed types. Mucinous and signet ring tumors tend to be the most malignant types. Few adenocarcinomas of the small or large intestine of mice and rats metastasize to the peritoneal cavity, lymph nodes, liver, or lung (Figure 25). Inflammation-induced or promoted colon adenocarcinomas tend to often be mucinous adenocarcinomas that form large cystic structures especially in the tunica muscularis and serosa (Figures 6, 26–30). Colon carcinogenesis may still follow the usual sequences of colon adenocarcinoma histopathogenesis (Cooper et al. 2000; Suzuki et al. 2004). They have been reported to form intraperitoneal tumor nodules on serosal surfaces, but metastases to lymph nodes, liver, and lung are less common (Erdman and Poutahidis 2010; Maggio-Price et al. 2005, 2006, 2009).
Other rodent intestinal tumors including sarcomas, GI stromal tumors, and lymphomas are described in the published nomenclatures but will not be discussed in this review (Whiteley et al. 1996; Nolte et al. 2014).
Inflammation and Colon Cancer
Chronic, smoldering, and polarized inflammation is associated with an increased risk of developing neoplasia (Balkwill, Charles, and Mantovani 2005; Coussens and Werb 2002). Numerous human cancers are associated with chronic inflammation caused by infections (i.e., Helicobacters [Polk and Peek 2010]) or toxins (i.e., tobacco smoke [Smith, Perfetti, and King 2006]) and irritants (i.e., asbestos [Yang et al. 2010]). There is an increased risk of developing colorectal cancer in human patients with IBD and the risk of developing colitis-associated cancer is dependent upon the intensity and duration of inflammation (Bernstein et al. 2001) increasing the overall incidence of colorectal cancer in IBD patients to 10 to 40 times that of the general population (Bachwich, Lichtenstein, and Traber 1994). Crohn’s disease and ulcerative colitis are the two most common types of IBD; both are human conditions with chronic and relapsing inflammation of the GI tract. The etiology of IBDs is likely multifactorial—a combination of genetic and environmental factors—with increasing awareness for the role of the intestinal microbiota and intestinal homeostasis. While no one model effectively recapitulates the complex and heterogeneous clinicopathologic features of human IBD, each can provide a tool to dissect the molecular pathways underlying colitis-driven cancer and to develop and test novel therapeutics. Mouse models of colitis-associated cancers (Table 1) include genetic (spontaneous or genetically modified), immunological (e.g., CD45RBhigh T-cell transfer), chemical- (e.g., DSS), and bacteria-driven (e.g., Helicobacter) and are reviewed in various publications (Kanneganti, Mino-Kenudson, and Mizoguchi 2011; Koboziev et al. 2011; Mladenova and Kohonen-Corish 2012; Oshima and Oshima 2012; Ostanin et al. 2009; Perse and Cerar 2012; Tanaka 2012a; Wirtz and Neurath 2007). Acute and chronic colitis models include those with epithelial integrity defects (leaky guts) via chemical stripping of the epithelium (DSS) or via genetic defects (e.g., multiple drug resistant gene 1a/P-glycoprotein knockout mice). The role of the innate and adaptive immune response in IBD and colorectal cancer can be explored using genetically modified mice (e.g., Il10 knockout [KO] and Rag2 KO) and immunodeficient mice adoptively transferred with colitogenic T-cells (colitis-inducing memory CD4+ T cells created at the time of disease development). Model selection will depend on the particular research question and, as with any mouse model, the effect of background strain; environmental factors and microflora on phenotype cannot be overstated. For example, C3H/HeJ mice are susceptible to DSS-induced colitis while C57BL/6J mice are partially resistant (Mahler et al. 1998, 1999). Multiple drug resistant gene 1a/P-glycoprotein knockout mice were initially developed on a Friend Virus B (FVB) background (FVB.129P2-Abcb1atm1Bor) and develop spontaneous colitis by 12 weeks (Wilk, Bilsborough, and Viney 2005) whereas on a C57Bl6/J background they are protected from spontaneous colitis (Staley, Schoeb, and Lorenz 2009). Colitis in the face of Il10 deficiency is also modified by background strains; severe disease occurs in 129SvEv background with decreasing severity in BALB/c and least severe in C57Bl/6 backgrounds (Berg et al. 1996). These inbred lines are useful tools to aid in characterizing genes that contribute to the relative disease susceptibility or resistance (Farmer et al. 2001; Mahler et al. 1999).
Selected mouse models of colitis-driven colorectal cancer by defect location.
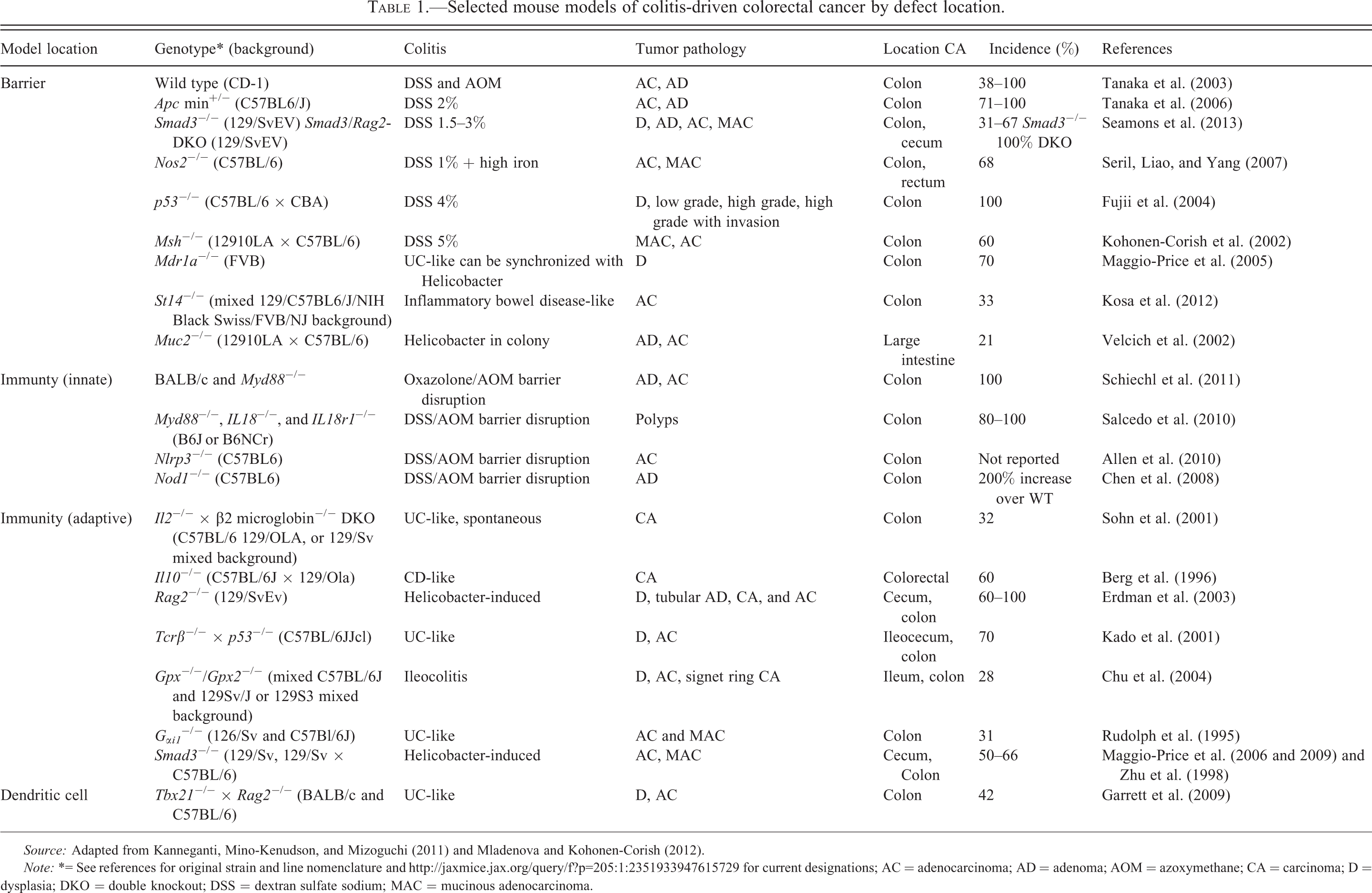
Source: Adapted from Kanneganti, Mino-Kenudson, and Mizoguchi (2011) and Mladenova and Kohonen-Corish (2012).
Note: *= See references for original strain and line nomenclature and http://jaxmice.jax.org/query/f?p=205:1:2351933947615729 for current designations; AC = adenocarcinoma; AD = adenoma; AOM = azoxymethane; CA = carcinoma; D = dysplasia; DKO = double knockout; DSS = dextran sulfate sodium; MAC = mucinous adenocarcinoma.
The influence of the microflora on colitis and colon cancer phenotype is demonstrated in a number of models including the Il10 KO as resident enteric bacteria are required to develop colitis (Sellon et al. 1998). The utility of enteric bacteria as research tools is exemplified by Helicobacter sp. The enterohepatic Helicobacter hepaticus was first described in A/JCr mice with chronic hepatitis leading to hepatocellular cancer (Ward et al. 1994). Other Helicobacter sp. were first linked to colitis in Il10 −/− mice (Fox et al. 1999) when a novel Helicobacter sp. was isolated from colitic Il10 KO mice. Further studies with Helicobacter sp. examined the role of CD4(+)CD45RB(lo)CD25(+)-regulatory T cells in colitis-driven cancer by the use of Helicobacter-infected 129/SvEv Rag2-deficient (Erdman, Poutahidis, et al. 2003) and Il10–deficient mice (Erdman, Rao, et al. 2003). Helicobacter sp. induces a colitis and eventually cancer in these lines, which is ameliorated by transfer of anti-inflammatory Il10–competent regulatory T cells (Erdman and Poutahidis 2010; Erdman, Poutahidis, et al. 2003; Erdman, Rao, et al. 2003).
Further evidence for the importance of bacterial triggers of colitis in genetically susceptible hosts is provided by studies examining Helicobacter sp. infection in mice Smad3 mutants (Smad3exo2/exo2), a transforming growth factor β pathway-signaling molecule. These Smad3-deficient mice were initially reported to develop colon cancer (Zhu et al. 1998); however, the phenotype was lost when the mice were rederived into a Helicobacter-free environment (Maggio-Price et al. 2006). Helicobacter-free Smad3 −/− (129S2/SvPasIco-Madh3tm1Par/J) were experimentally infected Helicobacter species, and in 50 to 66% of the animals the colon cancer phenotype was restored (Maggio-Price et al. 2006). This model was extended by creating Smad3/Rag2-DKO mice by intercrossing Smad3 −/− mice with Rag2 −/− to examine the loss of Smad3 in lymphocytes (Maggio-Price et al. 2009). The data suggested that the increased colon cancer susceptibility was due to: (1) excessive inflammation after a bacterial trigger secondary to T-regulatory cell dysfunction; (2) increased expression of proinflammatory cytokines and enhanced nuclear factor-kappaB activation; and (3) increased expression of both pro-oncogenic and anti-apoptotic proteins.
The AOM/DSS model of colitis-associated cancer is the most frequently used in preclinical testing of chemopreventive or therapeutic compounds (reviewed in De Robertis et al. 2011; Tanaka 2012b). Compound classes include cyclooxygenase-2 (COX-2) inhibitors, peroxisome proliferator–activated receptors (PPAR), plant-derived compounds, and probiotics (Kim et al. 2010). Many compounds tested have anti-inflammatory activity with suppressed expression of COX-2, inducible nitric oxide synthase (iNOS), and inflammatory cytokines (Tanaka 2012b). Examples include citrus-derived auraptene and nobiletin (Krishnan et al. 2009), tricin (Oyama et al. 2009), astaxanthin (Yasui et al. 2011), COX-2 selective inhibitor nimesulide (Orii et al. 2003) and PPAR ligands (troglitazone and bezafibrate; Kohno et al. 2005), and iNOS selective inhibitors (Kohno et al. 2007) among others. Studies using the AOM/DSS model have also examined the efficacy of probiotics such as Bifidobacterium lactis (Kim et al. 2010) and vitamin D (Kikuchi et al. 2007).
Preclinical Models for Colon Cancer Prevention and Treatment
Several mouse models for colon cancer prevention and therapy have been developed, other than those noted above for mouse colon cancer and inflammation models. Cancer chemoprevention is a term coined by physician scientist Michael Sporn at the National Cancer Institute (NCI) in the late 1970s for prevention of the development of cancer as an end point in rodent carcinogenesis studies (Sporn et al. 1976; Steele and Lubet 2010; Steele et al. 2009; Young, Ordonez, and Clarke 2013). It can be accomplished by exposing rodents to a test chemopreventive chemical or dietary component and targeting early changes in cells after exposure to a carcinogen, at early histopathologic stages of cancer development or later stages of carcinogenesis (Table 2). A variety of agents have been tested for various cancer organ models by the NCI Cancer Chemoprevention program and many other investigators (Green and Hudson 2005; Pereira et al. 1996; Pettan-Brewer et al. 2011; Steele and Lubet 2010). Often, studies test the ability of a prospective anticancer agent in a model system but do not define the targeted stage of tumor development or even cell or molecular target. The early successful colon chemoprevention studies used Apc mutant mice with anti-inflammatory reagents (Jacoby et al. 1996, Jacoby, Seibert, et al. 2000; Jacoby, Cole, et al. 2000), which may have led to later and current human clinical trials, especially in high colon cancer risk polyposis patients (Rial et al. 2012). There has been much work with rapamycin targeting the mammalian target of rapamycin pathway in many mouse cancer organ models including the colon (Deming, Leystra, Nettekoven, et al. 2013). Anti-inflammatory compounds have been shown in mice to prevent induced inflammation and inflammation-associated colon cancer (Kanneganti, Mino-Kenudson, and Mizoguchi 2011). The mechanisms of inhibition of inflammation or colon tumorigenesis are often not determined. Thus, its relevance to human disease is not yet known.
Key elements for the use of animal preclinical/nonclinical models for cancer chemoprevention in drug development.

Source: Steele and Lubet (2010).
Besides human tumor xenografts in immunodeficient mice, injecting mouse or human colon adenocarcinoma cells into mouse colon by orthotopic methods have been more recently developed (Choi et al. 2012; Zigmond et al. 2011; Martin et al. 2013; Hoffman 1999). These models may have more relevance to human treatment protocols. Imaging protocols can follow tumor progression and regression in these models (Choi et al. 2012; Young et al. 2009).
Footnotes
Acknowledgments
Jerrold M. Ward thanks Dr. John Weisburger for getting him started in this important field of cancer research. We thank Drs. Lillian Maggio-Price and Susan Erdman for providing some of the slides for preparation of the histopathology figures and the UW Histology and Imaging Core for technical support. We greatly appreciate the assistance of Beth Mahler for figure editing.
Authors’ Note
We received permissions from the Nature Publishing Group for publication of Figure 5 from an earlier 1974 paper in Laboratory Investigation by Jerrold M. Ward.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Piper M. Treuting is supported in part by CCFA #1579 (Maggio-Price PI http://www.ccfa.org/science-and-professionals/research/grants-fellowships/), Broad Medical Research Foundation (Maggio-Price PI), and AICR 09A136-Rev (Maggio-Price PI http://www.aicr.org/research/grant/).