
Abstract
Recent developments in the treatment of seizure disorders have been focused on medications with novel mechanisms of action. One such medication is perampanel, a first-in-class, noncompetitive, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor antagonist recently approved for the adjunctive treatment of partial seizures in patients 12 years of age and older when other antiepileptic medications have failed. The drug has demonstrated efficacy in several clinical trials, and appears to exert its anti-seizure activity in a dose-dependent manner. The drug has a half-life of approximately 105 hours suggesting the medication might be a logical option for individuals who have difficulty with adherence to medications that require multiple daily doses. Because of changes in serum concentration when enzyme-inducing antiepileptic drugs are employed concurrently, escalations in the initial dose of perampanel are recommended. Adverse events reported with perampanel use tend to be mild to moderate. However, psychiatric side-effects, including hostility and aggressive behavior, have been noted in some patients resulting in the inclusion of a warning on the Food and Drug Administration (FDA) approved labeling.
Introduction
Among the common types of seizures, partial-onset seizures (also known as focal seizures) are frequently among the most treatment resistant. Symptoms and impact on quality of life are dependent upon multiple factors including the site of origin, which determines symptoms and potential for alterations in consciousness, and whether or not the seizure remains localized or becomes secondarily generalized (as approximately 60% of partial-onset seizures do). 1 Even with the employment of multiple antiepileptic drugs, approximately one in three patients will continue to have seizures.2,3
The arsenal for the treatment of partial seizures has increased substantially in recent years. Several medications are approved by the Food and Drug Administration (FDA) for initiation of treatment as monotherapies, including the first-generation agents phenytoin, valproic acid, carbamazepine, phenobarbital. Newer (second-generation) agents approved for monotherapy include oxcarbazepine, topiramate, felbamate and most recently, lacosamide. Lamotrigine is also approved as monotherapy, but only when converting from carbamazepine, phenytoin, phenobarbital, primidone, or valproic acid. The list of drugs approved for adjunctive therapy is even more extensive, and includes gabapentin, pregabalin, tiagabine, levetiracetam, zonisamide, vigabatrin, ezogabine, and eslicarbazepine.
Traditional therapeutic targets for antiepileptic medications include voltage-gated ion channels (sodium and calcium in particular), or the augmentation of γ-aminobutyric acid (GABA). 4 More recently, efforts have been directed toward the development of medications with novel mechanisms of action. Glutamate, being the primary excitatory neurotrans-mitter in the brain, would seem to be a logical target for new moieties. It is well accepted that elevated concentrations of glutamate may trigger seizure activity, and higher than normal concentrations have been noted in the hippocampus both preceding and after the cessation of seizures.5,6 Two medications, felbamate and topiramate, possess the ability to bind to glutamate receptors (NMDA and kainite receptors, respectively). Activity at these sites is believed to contribute to the efficacy of these agents, though their primary mechanisms of action are focused elsewhere.
A third type of glutamate receptor is the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor. In addition to binding glutamate, this receptor type is a ligand-gated cation channel. 7 The greater the number of sites on an AMPA receptor that are occupied, the faster the current speed through the cation channel that opens in response to that binding.8,9 Because of the rapidity with which the channels open and close, it is believed that the AMPA receptors are responsible for the majority of excitatory transmission in the central nervous system. In the presence of heightened stimulation by glutamate, excessive calcium concentrations in cells may result. This in turn may lead to excitotoxicity.10,11
Perampanel
Perampanel (Fycompa®) is a first-in-class antiepileptic agent for the adjunctive treatment of partial seizures with or without secondary generalization in patients 12 years of age and older. It received approval for marketing from the European Commission in July, 2012. FDA approval was granted in October the same year.
Mechanism of action
Perampanel selectively binds to AMPA glutamate receptors and acts as a noncompetitive antagonist unable to be displaced by glutamate, even at excessive concentrations. 12 The drug was deliberately engineered to affect known mechanisms of seizure initiation and proliferation. 13 In the presence of perampanel, glutamate is still capable of binding to the AMPA receptor, but the anticipated response is inhibited by the presence of the drug at a separate binding site. It is believed that via this inhibition of glutamate receptor stimulation, seizure activity is suppressed. In a rat amygdala kindling model that introduced a higher intensity stimulus compared to the afterdischarge threshold, perampanel use resulted in a decrease in seizure severity, whereas other antiepileptic agents (carbamazepine, lamotrigine, levetiracetam and valproic acid) did not. 14
Pharmacokinetics and drug interactions
Perampanel displays a linear, one-compartment pharmacokinetic profile with first-order elimination.15–17 The drug is approximately 95% bound to serum proteins. After perampanel consumption, serum concentrations reach their maximum rapidly. The presence of food does not appear to change the extent of absorption, but time to achieve maximum serum concentration after a meal may be extended from approximately one hour to two or three hours.18,19 Once-daily dosing is possible because of the extended half-life of perampanel (52–129 hours after a single dose). 20 A pooled analysis of phase I trials demonstrated a half-life of 105 hours. 19 Using pharmacokinetic simulations derived from phase I trial data generated from 606 individuals, it has been demonstrated that missing a dose of perampanel, taking the missed dose within 12 hours, and then subsequently resuming the initial schedule results in no spike in plasma concentration. 21 The same analysis evaluated twice-daily dosing to determine if an evening-out of the time–concentration profile occurs in comparison to once-daily dosing. Though a 20% decrease in the fluctuation index was noted with multiple daily doses, the clinical significance of the change is expected to be minimal. However, individuals using enzyme-inducing antiepileptic agents concomitantly may experience more meaningful results depending on the extent of the interactions between the medications. 21
Metabolism of perampanel is extensive, and is carried out primarily by the cytochrome P-450 enzymes 3A4 and 3A5. This in turn is followed by glucuronidation. 19 As study data demonstrate that only 1/3 of a dose of perampanel is recoverable in the urine in healthy elderly volunteers (≥65 years of age), there is no recommendation for adjusting doses based on advanced age. Individuals with mild to moderate hepatic disease (Child–Pugh scores A or B) who are exposed to perampanel experience a decrease in clearance of approximately 50%. As such, dose adjustments are recommended with dysfunction at this level, and in the case of patients with more severe liver disease, the medication should be avoided entirely. 19 More specifically, the maximum recommended dose for individuals with mild hepatic impairment is 6 mg, and for moderate impairment is 4 mg. There are insufficient data to evaluate the safety of perampanel in individuals with severely compromised renal function or those undergoing hemodialysis. Subsequently, the drug should not be prescribed under these circumstances. 19
Drug interactions with medications that induce cytochrome P-450 enzymes have been shown to alter perampanel plasma concentrations. A decrease of as much as 67% has been demonstrated with concomitant therapy that includes enzyme-inducing antiepileptics (carbamazepine, oxcarbazepine and phenytoin). 19 For this reason, initial dosing at double the normal starting dose is recommended in the presence of these medications. Phenobarbital does not alter plasma concentrations of perampanel (though drug labeling suggests it is possible), and no other clinically significant interactions with other antiepileptic agents have been identified to date. Though the recommendation does not specify specific dosage changes, the manufacturer suggests that increased doses might be necessary when used in conjunction with medications and neutraceuticals known to induce hepatic enzymes (such as St. John's wort or rifampin). 19
Inhibitors of cytochrome P-450 enzyme metabolism may increase the likelihood of experiencing adverse events related to the use of perampanel because of higher than normal plasma concentrations. However, to date, no medication has been shown to increase concentrations of perampanel to this extent. The antifungal agent ketoconazole increases perampanel concentrations by approximately 20%. 19 In addition, perampanel may decrease the concentration of other medications, including levonorgestrel contraceptives, the levels of which have been shown to be lowered by 40% during high-dose (12 mg/day) perampanel administration. The use of a back-up birth control method when these two medications are used simultaneously is prudent. Specific dosing changes in relation to the simultaneous use of these agents have not been recommended, but it is important to note that interactions are possible.
Clinical efficacy trials
The data generated by three phase III clinical efficacy trials conducted in patients with partial seizures with or without secondary generalization led to the approval of perampanel as an adjunctive agent for the treatment of partial seizures (Table 1). The first of these was study 304, a high-dose, randomized, double-blind, placebo-controlled trial that included patients 12 years of age and older from several countries (Argentina, Canada, Chile, Mexico, and the USA). 22 The primary outcome measures for the study were 28 day seizure frequency compared to baseline and the number achieving a reduction in seizure occurrence of at least 50% (the responder rate). As would be expected, enrollees had epilepsy resistant to monotherapy, and were allowed to be on as many as three additional antiepileptic agents at stable doses. Three hundred eighty-seven patients were randomized in a 1:1:1 fashion to receive either placebo or active drug at a dose of 8 mg or 12 mg once daily. In order to meet eligibility requirements, the number of seizures experienced during the baseline phase (six weeks) had to total five or more per 28 days. The median number of seizures that patients reported during the baseline data collection period was 12–14.3 per 28 days. Target doses were achieved by titrating the drug upward over six weeks at 2 mg/week intervals, though dose decreases and longer titration schedules were allowed based on patient needs and tolerance of adverse events. Efficacy and safety data were collected four weeks beyond the 13-week treatment period.
Summary of phase III studies evaluating the efficacy of perampanel in patients with resistant partial-onset seizures.
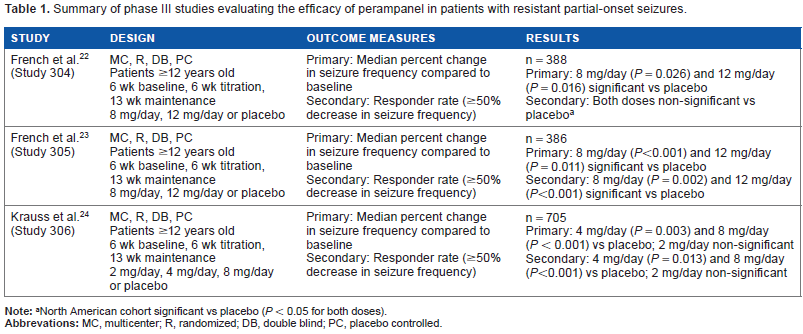
North American cohort significant vs placebo (P < 0.05 for both doses).
Both treatment groups achieved a significant decrease in the mean change in number of seizures (26.3% and 34.5% for the 8 mg and 12 mg doses, respectively). Individuals taking 8 mg daily demonstrated a median difference in seizure frequency of -13.5% compared to placebo. The 12 mg/day cohort demonstrated a difference of -14.2%. A 75–100% reduction in seizures was achieved by 18.8% and 17.3% of persons receiving perampanel 8 and 12 mg daily, respectively, in comparison to 5% receiving placebo (P = 0.001 for both doses). While no patient taking placebo achieved total seizure freedom, 2.2% of patients receiving 8 mg of perampanel daily and 1.5% of those receiving 12 mg daily did. The 50% responder rate for both the 8 mg (P = 0.076) and 12 mg/day (P = 0.091) groups did not differ statistically from placebo (37.6% [8 mg], 36.1% [12 mg] and 26.4% [placebo]). However, when individuals studied in North America were evaluated alone, the responder rate reached significance in perampanel-treated patients (P < 0.05 for both doses). No official explanation for the discrepancy in outcomes between the North American cohort and the Central and South American cohorts has been given, but speculation is that the differences may be the result of dissimilarities in methods of participant selection or investigator conduct.
A second high-dose phase III trial, study 305, included patients from Europe and Central Asia. The design of the study mirrored that of study 304. 23 The baseline median seizure frequency for the 386 enrolled patients, who were allowed up to three concomitant anti-seizure medications, was 11.8–13.7 per 28 days. During the double-blind phase of the study, the 8 mg and 12 mg daily dosing groups achieved median changes in seizure frequency of -30.5% (P < 0.001) and -17.6% (P = 0.011) vs baseline, respectively. Median seizure frequency was significantly lower for both doses vs placebo as well. For the 8 mg dose, the change was -19.1% (95% CI, -29.2% to -8.4%). The change for patients receiving 12 mg daily was -13.7% (95% CI, -25.2% to -2.3%). Achievement of a 75-100% decrease in seizure activity was seen in 15.5% and 16.5% of the patients taking 8 mg or 12 mg daily, respectively. Both decreases were significant in comparison to the change with placebo, which was 4.4%. While only 1.5% of patients receiving placebo were reported to have achieved total seizure freedom, patients given the 8 mg and 12 mg daily doses achieved seizure-free status at rates of 2.3% and 5%, respectively. The 50% responder rates for both the 8 mg dose (33.3%, P = 0.002) and the 12 mg dose (33.9%, P = 0.001) were significantly better than the rate demonstrated by the placebo group (14.7%).
The last of the phase III clinical trials, study 306, enrolled 705 patients, and was designed like the previous two studies. 24 However, lower doses of 2 mg and 4 mg were included with the highest dose of 8 mg daily. Enrollment occurred in Europe, Central Asia, Australia, and North America. Up to three concomitant anti-seizure medications were allowed per patient at stable doses. Baseline median seizure frequency was 9.3–10.9 per 28 days. The 2 mg daily cohort did not demonstrate a significant change in median frequency vs baseline. However, the 4 mg and 8 mg groups did with decreases of 23.3% (P = 0.03) and 30.8% (P < 0.001), respectively.
The 50% responder rate for placebo, 2 mg and 4 mg daily were 17.9%, 20.6%, and 28.5% (P = 0.013, number needed to treat = 8). The rate was higher for the 8 mg/day group at 34.9% (P < 0.001, number needed to treat = 6). Seizure-free status was not different between placebo and the 2 mg daily cohort (1.2% and 1.9%, respectively). Of patients in the 4 mg/day group, 4.4% were seizure free, and in the 8 mg/day group, 4.8% achieved seizure freedom.
Study 307 was a long-term extension study in which patients from the previously described three studies could be enrolled. 25 All subjects were given perampanel in this study, including those who had previously been randomized to receive placebo in another trial. Doses were titrated upward by 2 mg/day to a goal dose of 12 mg/day, or as high as the patient could tolerate if this dose was unachievable. The mean dose was 10.1 ± 2.3 mg/day. Open label dosing commenced up to a period of 256 weeks. Unlike during the phase III trials, dose adjustment of concomitant medications for epilepsy was allowed. Measured outcomes were consistent with phase III trial design, with assessment of efficacy being performed at 13-week intervals. Enrollment was robust with 96.4% of phase III trial participants continuing on in the extension. Prior to publication of the interim assessment data, 7.4% dropped out of the study because of a claim of lack of efficacy.
During the initial 26 weeks of the study, seizure rates dropped. From that point, frequency of seizure activity remained steady. For 588 patients with 12 months or more in the study, the most recent reporting of seizure activity prior to data analysis was a decrease of 47.2%. An additional analysis of 19 patients with more than 24 months of exposure revealed a 56% drop in seizure activity. Loss of efficacy in individual patients compared to data gathered during their enrollment in the previous trials was not assessed.
In taking the data generated from the phase III trials in total, the decrease in median seizure frequency was consistent. 26 Of the concomitant therapies taken by trial participants, carbamazepine, lamotrigine, levetiracetam, oxcarbazepine, and valproic acid were most frequently employed. Perampanel and levetiracetam co-administration resulted in a change of -39%, while use with carbamazepine produced a change of -18%. This begs the question as to whether any particular combination of perampanel and other antiepileptic agents has been shown to be more efficacious than others. To date, no specific rational combination therapy has been identified as it has been shown that the correlation between response and plasma concentration is maintained regardless of concomitant medication choices. 27
There does appear to be a dose–response relationship associated with perampanel use. Individuals in the phase III studies who received 8 mg/day and were given a final dose of 12 mg/day (n = 217) during the extension study conversion period had a median percent change in 28-day seizure frequency from -32.4% on the smaller dose to -44.9% on the larger one. 28 The responder rate increased from 37.3% to 42.9%. Considering the first 13 weeks of the maintenance period, the remaining 181 patients had a median decrease in seizure frequency of 11.9%, and an increase in responder rate of 7.2%. Those who were using the 12 mg dose in the phase III trials had seizure rates that were largely unchanged. 28 The optimal concentration of perampanel in the plasma has not been officially determined, though steady state concentrations of at least 70 ng/mL appear to be associated with better outcomes. 27
Safety and tolerability
The safety and tolerability of perampanel was studied in two phase II trials enrolling adult patients who had failed at least three antiepileptic medications. Study 206 enrolled patients who were randomized to 2 mg bid, 4 mg daily, or placebo. 29
Doses were adjusted at biweekly intervals over an eight-week time period following a four-week baseline period. Active drug was tolerated well regardless of schedule. Withdrawal rates were not significantly different in any of the groups (three, two, and one in the placebo, bid, and daily dosing groups, respectively). In the 153 patients included in the safety analysis, the rates of adverse events were similar between the placebo group (62.7%) and the active treatment groups (66.7%). Two out of four adverse events categorized as serious occurred in the placebo group. All serious events were seizure-related. Laboratory results did not reveal any concerns with active treatment. No deaths were reported.
For study 208, doses of up to 12 mg/day were administered to 48 individuals. 29 Stratification into the various cohorts (3:1 in favor of the active treatment group) was accomplished based on the presence of concomitant enzyme-inducing antiepileptic medication. Patients receiving active drug had their doses titrated upward by 2 mg/day every two weeks. Approximately 1/3 were able to tolerate the highest dose. In comparison, 60% of those taking placebo tolerated what represented the maximum dose for that group. Adverse events resulted in the withdrawal of one patient from the placebo group, and two from the active control group. Adverse events overall were similar between the treatment groups (80% and 84.2% for the placebo and active treatment groups, respectively). Serious adverse events were reported by one patient in each treatment group, but neither event was believed to be related to study treatment. There were no reported deaths or laboratory abnormalities described.
Overall, in the phase II trials, dizziness and somnolence were most commonly reported, and were observed in at least 10% of patients. Whether perampanel was responsible for these episodes is difficult to determine as both dizziness and somnolence are frequently associated with the antiepileptic drug class. As such, it is difficult to assign causality since the patients were on other concomitant medications for the treatment of seizures.
An extension trial including patients from studies 206 and 208 (study 207) resulted in preliminary results demonstrating mild to moderate adverse events in approximately 94% of those enrolled. 30 Seizure-related adverse events including convulsions, status epilepticus, and tonic–clonic seizures were deemed serious, and possibly related to drug therapy. Schizophrenia was also reported in a patient, and was possibly related to the study drug. One patient expired because of a myocardial infarction while in study 207, and though it is not believed that the event occurred as a result of perampanel use, sudden unexpected death in epilepsy (SUDEP) cannot be ruled out.
Safety data from study 307 (n = 1,216) have now been published. 31 A mean daily dose of 10.6 mg (±2.25 mg) was achieved by the 1,216 patients included in the analysis. The median exposure to perampanel was 1.5 years. The adverse events that emerged in at least 10% of those enrolled include dizziness, somnolence, headache, fatigue, irritability, and weight gain. Of those, only dizziness (3.9%) and irritability (1.3%) resulted in discontinuation. The two most commonly reported side-effects were dizziness and somnolence, except in North America where headache was the second most common complaint. Weight gain in adult patients averaged +1.9 kg at one year and +2.19 kg at two years. In adolescent patients, weight gain was more substantial with changes of +4.35 kg and +6.1 kg at one and two years, respectively. Serious adverse events that occurred in at least 1% of patients were exclusively seizure-related. A minimum of one adverse event was reported by 91.3% (n = 1,110) of enrollees. The majority (80.2%) were defined as either mild or moderate in intensity. Sixteen percent (n = 195) withdrew for reasons related to adverse events, while 39.7% required dose reductions, and another 3.9% required temporary interruption of perampanel intake. The side-effect profile described for study 307 is consistent with a meta-analysis of nine randomized, controlled trials (five conducted in patients with epilepsy and four in patients with a diagnosis of Parkinson's disease) that summarized the adverse event findings in approximately 4,000 patients, 2,627 of whom were given perampanel. 32 In those trials, dizziness, ataxia, somnolence, irritability, and increased weight were associated with perampanel use, while increased seizure activity was associated with placebo.
Considering psychiatric adverse events that emerged during study 307, the most common were depression (5.4%), insomnia (5.3%), aggression (5.1%), anxiety (5%), and mood swings not otherwise specified (2.1%). A total of 47 patients (3.9%) experienced at least one adverse event from the psychiatric domain. Ten individuals reported more than one. Of those individuals reporting any psychiatric adverse event, approximately 43% had a previously documented history of a psychiatric diagnosis. A breakdown of specific psychiatric events reveals that the most common was aggressive behavior (12 patients); followed by psychiatric disorder not otherwise specified and suicidal ideation (6 patients each); affective disorders, depression, and suicide attempt (4 patients each); paranoia (3 patients); and abnormal behavior, acute psychosis agitation, and disorientation (2 patients each). In 11 of the 12 individuals reported to have exhibited aggressive behavior, the side-effect resolved. Seven of these individuals withdrew from the study entirely, four were able to continue on a stable dose of perampanel, and one required a decrease in dose. There were no data on the 12th patient. As a result of the appearance of aggressive, angry, or hostile behavior with perampanel use, a boxed warning has been included in the approved labeling. 19 Close monitoring is recommended, and dose reduction is advised should such symptoms appear. In the case of severe symptoms, symptoms that do not respond to dose adjustment or worsening symptoms, the drug should be discontinued.
A series of case reports described a possible increased risk of suicidality in 3 of 23 patients treated with perampanel. 33 One of the three had a previous history of suicidality, and all three were described as having cognitive impairment of varying degrees. The patients were not receiving the same concomitant medications, so there was no suggestion that a specific drug combination was more likely to result in suicidal tendencies. No evidence of higher than expected serum drug levels was found. The authors claim to have observed suicidal impulses in two of the three patients that seemed to be associated with symptoms akin to irritability as opposed to depression. 33
In total, study 307 represented 1,803 patient-years of exposure to perampanel. Five deaths occurred over the duration of the study. However, none were believed to have transpired because of perampanel use. Three of the deaths were related to seizure activity (two experiencing SUDEP and one who sustained a head injury because of epileptic activity).
An analysis of pooled data from phase III perampanel studies revealed that treatment emergent adverse events (TEAE) were apparent in 294 patients receiving placebo (66.5%) and 799 patients receiving the active drug (77%). 34 The most common adverse events reported across the studies were dizziness, somnolence, fatigue, irritability, nausea, and falls. Rates of severe adverse events were similar when comparing the placebo and active treatment cohorts, with 5% of patients receiving placebo and 5.5% of patients receiving perampanel experiencing such events. TEAE categorized as severe included a fall, nephrolithiasis, cholecystitis, hemorrhagic cystitis, urinary incontinence, and thrombocytopenia. When considering data from all three studies, the withdrawal rate attributed to adverse events was 9.5%. There is a clear relationship between treatment discontinuation and perampanel dose. Of those patients receiving 4 mg/day, 2.9% discontinued therapy. In contrast, withdrawal rates for patients receiving 8 mg and 12 mg of perampanel daily were 7.7% and 19.2%, respectively. No clinically important changes in vital signs, ECG result, or hematological evaluation were noted in any treatment group.
Four individuals receiving placebo and 12 receiving perampanel experienced psychiatric TEAE. Aggression was most common and was more likely to occur in adolescent patients (7.8%) compared to adult patients (1.3%). The rate of aggression was similar between the 4 mg/day group and the placebo group (0.6% vs 0.5% respectively), but was noted to be higher in individuals receiving 12 mg/day (3.1%). Depression occurred in 0.6% of patients taking low dose (4 mg) perampanel and in 2.4% of those taking high dose (12 mg) perampanel. The rate reported in patients receiving placebo was 1.6%.
Though one patient was reported to have admitted to suicidal ideation, overall no increased risk was felt to be demonstrated by phase III data. 34 During ongoing monitoring, one case of deliberate perampanel overdose has been noted and described. 35 The individual in question, a 34-year-old female diagnosed as having tuberous sclerosis with complex focal and generalized seizures since the age of 15, but with a normal IQ, ingested 204 mg of perampanel at one time. Post-ingestion, the patient's Glasgow Coma Scale score was eight. Because of the rate at which perampanel is absorbed and distributed throughout the body tissues, no gastric lavage was employed. Impairment of consciousness continued for approximately two days during which no respiratory support was needed. At no point was epileptiform activity noted during EEG monitoring. The patient was also receiving levetiracetam, pregabalin, and topiramate concomitantly, and was reportedly taking them at stable doses.
Perampanel has been designated pregnancy category C, indicating that developmental toxicity has been observed in animal studies. Toxicity was observed in pregnant rats (increased visceral abnormalities, delayed sexual maturation, and pup deaths). In rabbits, no toxicity was noted with exposure to drug up to the equivalent of two times the 8 mg dose. 19 The manufacturer recommends avoidance in pregnancy if possible. In the case that therapy is to be continued during pregnancy, or if unintentional exposure during pregnancy occurs, patients may be enrolled in the North American Antiepileptic Drug Pregnancy Registry. It is unknown as to whether perampanel or its metabolites are excreted to a significant extent into human breast milk.
Summary
Perampanel is the first antiepileptic drug specifically designed to bind to AMPA receptors in an effort to offset the excitatory response to elevated glutamate concentrations believed to play a role in seizure propagation. It has demonstrated efficacy as an adjunctive agent for patients with refractory partial seizures, and its action is believed to be at least as robust as other antiepileptic drugs that share this indication. Perampanel's pharmacokinetic profile is straightforward, and the drug's long half-life may be advantageous for patients who experience difficulty with adherence. The drug is well tolerated overall, with most reported adverse events described as mild or moderate in nature. The FDA approved package insert for the drug does contain a boxed warning describing aggression, hostility, irritability, anger, and homicidal ideation and threats as adverse reactions associated with perampanel use that warrant increased vigilance. The starting dose (in the absence of enzyme inducers) is 2 mg, and the drug may be titrated in 2 mg increments each week to a target of 8–12 mg per day. Dosing should be individualized based on concomitant therapy, specifically the presence of co-administered enzyme-inducing antiepileptic agents, which requires the initial dose to be doubled to 4 mg daily at therapy outset.
Author Contributions
Conceived the concepts: MF. Wrote the first draft of the manuscript: MF. Made critical revisions: MF. The author reviewed and approved of the final manuscript.