
Abstract
Castleman's disease (CD) is a group of rare lymphoproliferative disorders sharing characteristic clinical and histological features, and usually accompanied by a marked systemic inflammatory response. Three histological patterns of lymph nodes were described: the hyaline-vascular, the plasma-cell and the mixed types. The former is more common (80%-90%) and tends to be localized. The plasma cell type is more aggressive and usually multicentric. It is interesting that the inflammatory manifestations seem to be related to a lymph node lesion, because the systemic symptoms and inflammatory activity can return to normal after surgical excision or successful medical treatment of the disease.
We report here our 15-year experience with this rare disease in King Fahd Hospital of the University, Al-Khobar, Saudi Arabia, focusing on the clinical features, therapy, and patients'outcome.
Keywords
Introduction
Castleman and Towne 1 described a disease presenting as a mediastinal mass resembling thymoma. It is also known as “giant lymph node hyperplasia”, “lymph node hamartoma”, “angiofollicular mediastinal lymph node hyperplasia”, and “angiomatous lymphoid hyperplasia”. The pathogenesis is unknown, but the bulk of evidence points toward faulty immune regulation, resulting in excessive B-lymphocyte and plasma-cell proliferation in lymphatic tissue. In addition to the mediastinal presentation, extrathoracic involvement in the neck, axilla, mesentery, pelvis, pancreas, adrenal gland, and retroperitoneum also have been described.2–7 There are 3 major pathologic variations of Castleman's disease 8 : (1) hyaline-vascular type, the most frequent, characterized by small hyaline-vascular follicles and capillary proliferation; (2) the plasma-cell type, in which large lymphoid follicles are separated by sheets of plasma cells; and (3) less often a combination of both types (the mixed type) may occur. The hyaline-vascular cases usually are largely asymptomatic, whereas the less common plasma-cell variant may present with fever, anemia, weight loss, and night sweats, along with polyclonal hypergamma-globulinemia. The diagnosis of Castleman's disease is ultimately by histology, thereby requiring either removal or biopsy of the lesion for definitive diagnosis. The hyaline vascular variant is characterized by extensive capillary proliferation and a lymphocyte-predominant infiltrate surrounding small germinal centers. Hyalinized fibrous tissue surrounding the proliferating capillaries is typically present.9,10 The plasma cell variant has sheets of mature plasma cells within the interfollicular tissues surrounding larger germinal centers and has significantly less vascularity. 10 The localized form may originate from either of these variants, but the multicentric version is almost exclusively derived from the plasma cell variant. In addition, there are some benign and malignant conditions, including lymphoma and thymoma, that may appear histologically similar to Castleman's disease. Therefore, immunohistologic and immunologic gene rearrangement studies of the specimens can be useful in solidifying the diagnosis. Identifying an immunophenotypically varied population of B lymphocytes with polyclonal surface and cytoplasmic immunoglobulin markers helps to confirm the diagnosis of Castleman's disease and differentiate it from lymphoma. 11
Castleman's disease is a rare lymphoproliferative disorders. Few cases have been described world widely. We are describing 8 cases of Castleman's disease which have been diagnosed over the last 15 years at King Fahd Hospital of the University in Saudi Arabia.
Material and Methods
Patients
This is a retrospective study, in which the records of all patients who had lymph node resection at King Fahd Hospital of the University, for the last 15 years were studied. All cases with pathological findings consistent with CD of the hyaline vascular or the plasma cell variant were included in this study. An independent pathologist examined the histologic features of the lymph nodes and/or other tissues collected from the patients and confirmed the histologic diagnosis. None of the patients had autoimmune diseases such as rheumatoid arthritis or Sjögren syndrome.
Serum Protein assays
Serum concentrations of C-reactive protein (CRP) and serum amyloid A protein (if needed) were measured by commercial automated immunoassays with lower detection limits of 0.2 mg/l and 0.7 mg/l respectively.
Il-6 values
Were measured by commercial enzyme immunoassay (Biosource International). Values above 4 pg/ml are considered abnormal.
How Lymphoma Was Ruled out?
Having any doubts about the diagnosis, the following have been used on different tissue samples (including bone marrow) to rule out lymphomas:
Cytomorphology assessed on Papanicolaou stain (alcohol-fixed) and Diff-Quik (air dried) smears.
Immunophenotyping by flow cytometry or immunocytochemistry on cytospin Preparations.
Immunostaining with Ki-67.
Statistical analysis
Only simple tests (average, mean, percentages) are used because of the small number of patients due to rarity of cases.
Results
Case histories
Patient 1
A 24-year old female patient was referred to us because of 6-month history of unilateral neck swellings. She was otherwise healthy. The patient asked for medical advice because of cosmetic reason. Physical examination was normal apart from enlarged right lower deep cervical lymph nodes. The lymph nodes were discrete, firm, mobile, non-tender, and not attached to the skin or underlying structures. There were no other palpable lymph nodes. Abdominal, chest, cardiac, and neurological examinations were normal. Laboratory investigations are shown in Tables 1 and 2. Chest radiograph and thorax tomography were normal as well as abdominal ultrasound. One cervical lymph node was removed. In gross appearance, the surface of the mass was pinkish gray and well encapsulated. The histopathologic examination showed diffuse follicular appearance with the characteristic features of Castleman's disease. Lymphoid follicles contained vascular structures in their germinal centers with hyaline-like changes. There were no plasma cells in the interfollicular areas. The picture was consistent with the hyaline vascular type of Castleman's disease (Fig. 1).

CD: hyaline vascular type. Lymphoid follicles stained with H and E, contained vascular structures in their germinal centers with hyaline-like changes. No plasma cells in the inter-follicular areas. Original magnification x 200.
Demographic and clinical characteristics of patients.

Abbreviations: C, cervical; Ms, mesenteric; A, axillary; Md, mediastinal; I, inguinal; HV, hyaline vascular; PC, plasma cell type; LN, lymphadenopathy; NA, not available; Skin, skin rash; MCG, monoclonal gammopathy.
Demographic and clinical characteristics of patients (Cont.).

Abbreviations: NR, non reactive; HBsAg, hepatitis B surface antigen; HCVAb, hepatitis C virus antibodies; HIV, human immune deficiency virus antibodies; CMV, cytomegalovirus antibodies; PPD, purified protein derivative (tuberculin test).
Patient 2
A 52 year-old male patient presented with abdominal discomfort and four-month history of diarrhea. Physical examination showed non-tender central intraabdominal mass. The laboratory results are presented in Tables 1 and 2. Abdominal ultrasonography examination and computed CT scan (Fig. 2) depicted a heterogeneous solid mass measuring 5.8 x 5.0 cm with no organ enlargement. Chest radiograph and tomography were normal.
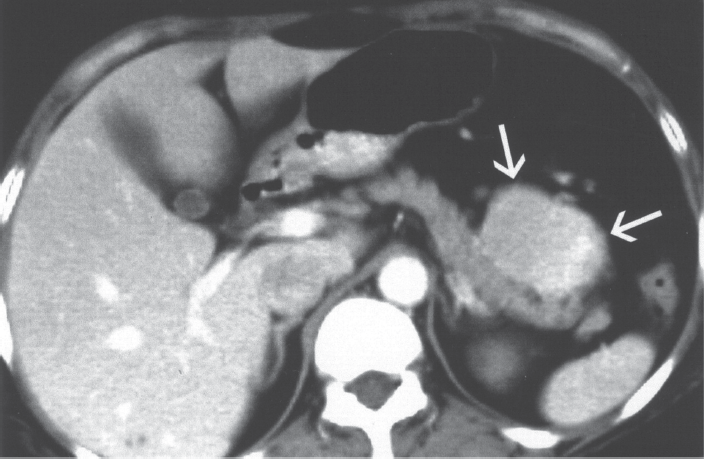
CT scan of the abdomen in a patient wit CD showing heterogenous solid mass with no organ enlargement. At laparoscopy the tumor was found to arise from the mesenteric lymph nodes.
At laparoscopy the tumor was found to arise from the mesenteric lymph node group and was not attached to any organs. It was resected easily. Histologic examination yielded the diagnosis of Castleman's disease; the hyaline-vascular type.
The patient's recovery was uncomplicated, and he remains healthy and free of disease 4 years later.
Patient 3
A 56-year old Indian professor presented with 5-months history of fever spikes, abdominal pain, loss of appetite and weight. During hospital stay he developed progressively increasing jaundice and abdominal distension. Physical examination showed ill looking man who was also pale and jaundiced. A tiny lymph node was palpated in the right supraclavicular group which was unapproachable to surgeons. Chest examination showed right-side pleural effusion, while cardiac examination was normal. Abdominal examination revealed hepatosplenomegaly and ascites. There was +3 pretibial edema, and sensory polyneuropathy. Laboratory investigations are shown in Tables 1 and 2. Bone marrow biopsy was normal. The ascitic fluid was transudate with negative culture and negative cytology. Abdominal CT scan showed hepatosplenoegaly, and multiple mesenteric lymph nodes. At laparotomy multiple mesenteric and celiac lymph nodes were found; some of them were compressing the common bile duct; biopsies were taken. Histologic examination yielded the diagnosis of Castleman's disease; the plasma cell disseminated type (Fig. 3). The patient was treated with systemic steroids and cyclophosphamide, but he died after two weeks from onset of therapy because of disseminated intravascular coagulopathy.

CD, plasma cell disseminated type. H and E stain. Original magnification x 100.
Patient 4
A 47-year old male patient was admitted to the hospital because of dyspnea on exertion, dry cough and fever for three months. He was seen in another hospital and treated with different antibiotics and bronchodilators; his symptoms improved. Later on, cough and fever recurred with aggravation of dyspnea and wheezing. In addition he noticed erythematous skin rash all over his upper and lower limbs. Initially the skin rash was interpreted as drug eruptions, treated with antihistaminics without improvement. He was then referred to our hospital. Upon examination, temperature ranged from 38.0-39.5 °C. Macular skin rash was found on both upper and lower limbs. Right upper deep cervical and bilateral axillary lymph nodes, which were firm, mobile and non-tender. Jugular venous pressure was normal. Chest examination showed diffuse bilateral expiratory ronchi, while cardiac examination was normal. The abdomen was soft and lax with palpable liver and spleen. The remainder of the examination was normal. Tables 1 and 2 show the laboratory investigations. CT scanning of the chest, abdomen and pelvis showed multiple nodular opacities in both hilar areas, multiple enlarged lymph nodes in the left para-aortic region and in the peripancreatic space; in addition both the liver and spleen were enlarged with nodular splenic calcifications. Bone marrow aspirations and biopsies were negative for acid fast bacilli, bacterial cultures, fungi, granulomas or malignancies; it was otherwise hypercellular (Fig. 4). Two cervical lymph nodes were totally removed and examined; many of the lymphoid follicles displayed atrophic germinal centers, and mantle zone expanded by large plasma cells (Fig. 5). These cells expressed monotypic IgM lambda on both immunohistochemical analysis and in situ hybridization. These features are characteristic of the multicentric Castleman's disease (MCD).
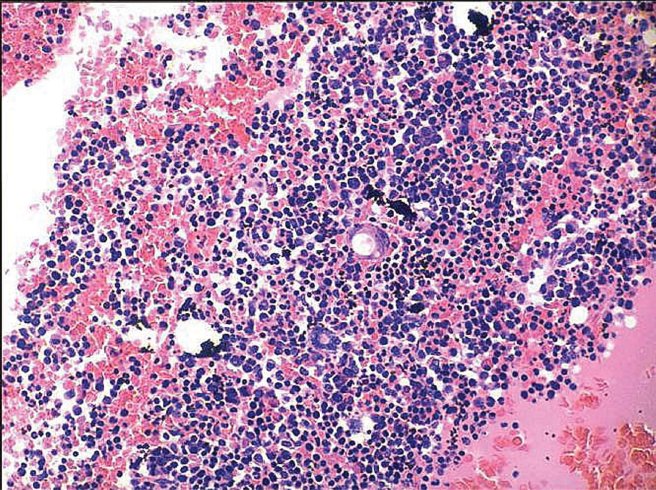
Bone marrow biopsy in a patient with CD showing hypercellular bone marrow. H & E stain. Original magnification x 100.

Lymph node biopsy in multicentric CD with atrophic germinal center and mantle zone expanded by large plasma cells. H & E stain. Original magnification x 100.
The patient was treated with high dose of intravenous methylprednisolone plus anti-CD20 monoclonal antibody rituximab (Rituxan; Hoffman-La Roche, Basel, Switzerland). The liver size regressed to normal, fever disappeared, and chest condition improved, but splenomegaly persisted. He is still on regular follow-up.
Patient 5
A 34-year male patient with a history of an axillary swelling for at least 2 years, presented to our hospital with postural dizziness, facial and lower limbs edema, in addition to dyspnea on exertion. Upon examination, he looked pale, with eyelid swellings, but no jaundice or cyanosis. Supine blood pressure was 144/86, and standing 110/64 mmHg. Internal jugular venous pressure was 12 cm H2O. Multiple lymph nodes were palpable in the left axillary area; they were firm, mobile, smooth, discrete, and non-tender. Other lymph nodes could not be palpated. Chest examination showed bilateral pleural effusion, and cardiac evaluation was normal. Abdominal examination was normal except for ascites. The kidneys were not palpable. Musculoskeletal system including all the joints was free. There was 4+ pitting edema of both lower limbs, the distal pulsations were all intact. Tables 1 and 2 show the laboratory investigations. Chest X ray, normal cardiac shadow with bilateral pleural effusion; ECHO cardiography, normal sized cardiac chambers and valves with normal fractional shortening and no pericardial effusion; serum iron, TIBC, and serum ferritin were normal; abdominal ultrasonography revealed normal size liver and spleen but bulky kidneys on both sides. The pleural and ascetic fluids were transudative with negative bacterial and fungal cultures. All the right axillary lymph nodes were removed and examined; it was interpreted as Castleman's disease, mixed type. Kidney biopsy was performed under local anesthesia; the renal tissue was examined histologically and it showed amyloid deposits (Fig. 6). The diagnosis of AA-amyloid type was confirmed. IV methylprednisolone and IV cyclophosphamide were started.

Kidney biopsy stained with Congo red in a patient with CD complicated by amyloidosis. Original magnification x 200.
Follow-up: two weeks following lymph node excision and the IV medications; lower limb and facial edema subsided. Ascites and pleural effusion disappeared in two month-time. Repeated 24 h urinary protein was 3.1 gm. The patient was followed-up on no treatment. Throughout the 7-years follow-up, he has no complaint, renal functions were normal, and 24-h urinary protein ≤300 mg.
Patient 6
17-year schoolgirl was seen in the outpatient department with 11-months history of left cervical swellings, and a 3-months history of fever, loss of appetite and weight. She had no respiratory, cardiac, gastrointestinal, or central nervous manifestations. There was no history of contact with infected patients, raw milk ingestion or traveling abroad. On examination, she is pale but not jaundiced. Temperature was 39.8 °C, blood pressure 110/70 mmHg and respiratory rate 16/min. Head and neck examination was normal except for bilateral cervical (submandibular, submental, upper and lower deep cervical) lymph nodes; the biggest was 4 x 3 cm, and the smallest was 1 x 0.5 cm. All the lymph nodes were discrete, firm, easily mobile, non tender, with smooth surface and normal skin overlying it. Chest, heart, musculoskeletal, and CNS examination were all normal. The abdomen was lax, freely mobile and non-tender; splenomegaly; no ascites or abdominal bruit. Investigations are shown in Tables 1 and 2. chest radiogram, abdominal, chest and pelvic CT scan were all normal. Cervical lymph node biopsies (most of the lymph nodes were removed) and histological examination yielded the diagnosis of Castleman's disease; the hyaline vascular type.
Patient 7
A 44-year old nurse had nearly 18 months history of cervical and axillary swellings, night fever, night sweating, loss of appetite, easy fatigability and skin rash over the upper limbs and chest. She was previously treated with empirical anti-TB medications for more than three months (she refused lymph node biopsy) with no response. Once she was tried on antiviral medications because of positive CMV-antibodies (of the IgG type); fever and lymph node enlargement, however did not improve. Upon examination; temperature was always above 38 °C; erythematous skin rash allover the upper limbs chest and back. No mouth, tongue or pharyngeal ulcers, although the tongue was coated with what it seemed to be candida and the pharynx was significantly congested. Right metatarsophalyngeal joint of the big toe had evidence of gouty arthritis. The right cervical and both axillary lymph nodes were enlarged; discrete, non-tender, firm and mobile. Chest, heart and abdominal examinations were all normal as well as both breasts. She was thoroughly investigated for a cause of her problems and all we got was a positive throat and nasal swab cultures for MRSA that was sensitive only to vancomycin. Other laboratory investigations are shown in Tables 1 and 2. The dermatologist believed that the skin rash a drug eruption. Skin biopsy confirmed his suspicion, and it turned out that she was using some herbal medication which was stopped upon our advice; the rash gradually fades out. She had axillary lymph node resection. Histological examination yielded the diagnosis of Castleman's disease; the hyaline vascular type.
Patient 8
A 39-year-old diabetic man, a known case of status post kidney transplantation and long-standing left diabetic foot and diabetic neuropathy, presented with progressive shortness of breath, cough, and expectoration of yellow/green sputum for two weeks prior to admission. He had on and off fever for the last 4 months for which he used to be admitted to hospital with bilateral pulmonary infiltrates. Being heavily immunocompromised; he received broad spectrum antibacterial and antiviral medications, with partial remission. A definite micro-organism, however, could not be identified. He had significant inguinal lymph node enlargement for over 20 months; that was always attributed to the left foot infection. During the last admission we noticed that the inguinal lymph nodes got bigger and that there appeared to be a small right supraclavicular lymph node which was 0.5 x 0.5 cm, firm, mobile, smooth, and non-tender. In addition abdominal examination yielded splenomegaly; about 5 cm below the left costal margin, firm and non-tender, the liver span was 11 cm, and there was no ascites. Both hepatitis C and B screenings were negative, as well as HIV testing. ANA was 1:640, rheumatoid factor was positive, Cryoglobulins and other antibodies were not found. Cultures from different places including those from the foot were negative, as well as virology and mycology studies. The results of the other laboratory investigations are shown in Tables 1 and 2. Chest X-ray revealed bilateral pulmonary infiltrates and chest CT scan added bilateral mediastinal shadows that were interpreted by the radiologist as mediastinal lymph nodes. Echo cardiography was normal apart from pericardial effusion. Bronchoscopy and broncho-alveolar lavage were negative for malignancy, AFB, fungus or pneumocystis carinii. Abdominal and pelvic CT scans were negative. Supraclavicular lymph node biopsy showed findings consistent with Castleman's disease; the plasma cell type. The patient was treated with high dose of intravenous methylprednisolone plus anti-CD20 monoclonal antibody rituximab (Rituxan; Hoffman-La Roche, Basel, Switzerland). Fever disappeared and the lung infiltrates resolved within 10-day period.
Evaluation of cases
Tables 1 and 2 show the demographic and clinical characteristics of patients. Three (37.5%) patients were females and 5 (62.5%) were males. The average age is 37 years (range 18-56). The mean duration of disease is 12 ∓ 8 (range 4-24) months. The sites of lymph nodes are as follows: 62.5% cervical, 25% mesenteric and other intra-abdominal, 25% mediastinal, 25% axillary, and 12.5% inguinal. Anemia (defined as hemoglobin ≤12.0 gm/dl) was present in all (100%) patients, 87.5% had hypoalbuminemia (serum albumin ≤3.5 g/dl), renal impairment (defined as serum creatinine ≥ 1.2 mg/dl) was present in 50%, polyclonal gammopathy in 65% and monoclonal gammopathy in 12.5%, and AA-amyloidosis in 12.5%. All patients (100%) tested for IL-6 had elevated blood values with a range of 11.8-176.0 pg/ml. As for the histological subtypes; 3 (37.5%) patients had the hyaline vascular, 4 (50%) plasma cell type, and 1 (12.5%) mixed.
Table 3 demonstrates the type of treatment and the patients’ outcome. One patient (12.5) died during the study period; the cause of death was DIC (disseminated intravascular coagulopathy), 6 (75%) had complete recovery after lymph node resection. Rituximab and chemotherapy were used in 2 (25%) patients with complete response.
Treatment and outcome of the 8 cases with Castleman's disease.

Discussion
Angiofollicular lymph node hyperplasia or Castleman's disease (CD) was first described by Castleman and Towne in 1956. 12 It is an uncommon clinicopathologic entity which traditionally has been divided into hyaline vascular and plasma cell types based on histologic differences. 13 Castleman's disease has two types of clinical presentations (localized and multicentric) and three histologic subtypes (hyaline vascular, plasma cell and mixed).14,15 The plasma cell variant is associated with a variety of unusual syndromes, including myasthenia gravis, nephritic syndrome, peripheral neuropathy, amyloidosis and temporal arteritis. 16 The plasma cell variant tends to be a local form, which occurs more commonly in young adults and is amenable to local therapy.17,18 The multicentric form occurs in older adults or in HIV infected patients with diffuse adenopathy and a variable, but often fatal clinical course in which 20 to 30% of patients develop Kaposi sarcoma or B-cell lymphoma.19,20 Immunophenotyping and IgG gene rearrangements of the plasma cell variant in CT indicate that patients usually have polyclonal lesions. However, monoclonal Ig gene rearrangements have been identified in patients with the multicentric form and patients who develop B-cell lymphoma.21,22 KSHV (Kaposi's sarcoma H virus) (HHV8) has been identified in HIV associated multicentric CD cases. KSHV positive plasmablasts have lambda light-chain restriction and are localized to the mantle zone of germinal centers. They can coalesce to form microscopic lymphomas. The lymphomas have had a variable histologic pattern but they are usually of B cell origin with mantle cell being the most common subtype and have a poor prognosis. Increased expression of the gene encoding for IL-6 has been identified in CD, and retroviral transduction of the gene into mice has reproduced histology and symptoms. Moreover antibodies to IL-6 or its receptor have ameliorated the disease.23–25 From a clinical staging point of view, Castleman's disease is classified into a localized form, and a more generalized lymphoproliferative disorder with more extensive lymph node involvement and severe systemic manifestations referred to as disseminated/multicenteric subgroup with or without peripheral neuropathy.26–30 This division is not absolute as mild peripheral adenopathy and splenomegaly may even be seen in the localized form. The etiology of the disease is unknown, however, a viral etiology resulting in disordered immunoregulation and dysplastic lymphoproliferative process has been postulated.26–28 The clinical presentations of the two types differ with the localized form having a benign outcome, and the disseminated form following a more dire course associated with organomegaly, polyneuropathy, concurrent endocrine gland disease, proteinuria, infections, and skin changes. Various cytokines and growth factors are implicated in the pathology of the disseminated type. Interleukin 6 (IL-6) and vascular endothelial growth factor (VEGF) are the most important among them. Yoshizaki et al. 33 noted elevated serum IL-6 concentrations in patients with Castleman's disease that decreased to normal values after lymph node masses had been resected. IL-6 is a pleiotropic cytokine with a wide range of biological activities. It causes proliferation and differentiation of B cells into antibody producing cells, resulting in hyperplastic follicles and lymph node enlargement. IL-6 also increases VEGF secretion, causing angiogenesis, proliferation of vascular muscle cells, and capillary proliferation with endothelial hyperplasia. IL-6 also is responsible for polarization of T lymphocytes to a type 2 cytokine profile leading to autoimmune phenomena, including autoimmune hemolytic anemia, antinuclear antibody positivity, and increased IgE levels. IL-6 induces synthesis of hepatic acute phase reactants and increases the erythrocyte sedimentation rate and C-reactive protein, IgG, serum fibrinogen, and serum amyloid-A protein levels. Systemic manifestations virtually are always associated with increased IL-6 levels.34,35 VEGF is expressed strongly in plasma cells in the interfollicular region of the lymph nodes. It can influence and modify the activity of CD and systemic involvement. Autocrine action of VEGF from plasma cells may be responsible for pleural effusion, ascites, edema and proteinuria. 36 It acts by enhancing vascular permeability and was reported to protect and accelerate renal recovery in antiglomerular endothelial antibody-induced thrombotic microangiopathy in rats. Thus, the role of VEGF, whether protective or causative in systemic injury is not precisely known. 37 In the present study, serum IL-6 and VEGF levels were not studied. Other cytokines such as IL-1β and IL-1α may also play a role. 36 Recently Kaposi's sarcoma associated herpes virus (KSHV or HHV-8) which encodes a functional cytokine (vIL-6) has been found in some patients with Castleman's disease suggesting that aberrant IL-6 activity can result from either endogenous or viral sources.38,39 The human herpes virus 8 (HHV-8) can be associated with Kaposi's sarcoma and primary effusion lymphoma. 39 It is found in the lymph nodes of a high proportion of patients with multicentric CD. IL-6 is present at elevated levels in Kaposi's sarcoma and multicentric CD lesions, and IL-6 promotes the growth of Kaposi's sarcoma cells in culture. HHV-8 encodes a homolog of human IL-6 that is known as viral IL-6 (vIL-6), which acts as mitogenic factor and induces intracellular signaling that is generally the same as IL-6.24,25,39,40 CD, the multicentric form may be associated with POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome, 41 a multisystemic syndrome associated with plasma cell dyscrasia. In both CD and POEMS syndrome, various cytokines and growth factors are implicated in the pathogenesis of the disease and may be responsible for abnormalities like hypergammglobulinemia, thrombocytopenia, and infiltration of inflammatory cells into the tissues, in addition to splenomegaly, and lymphadenopathy. 42 Mylona et al reviewed 84 HIV positive patients. They had polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS) syndrome which was seen in approximately 15% of HIV negative individuals with MCD, specially those infected by HHV-8. 43 One of our patients (patient number 3) had hepatosplenoegaly, polyneuropathy, skin changes and thrombocytopenia; however endocrinopathy and monoclonal protein pattern were not seen. Hemophagocytic syndrome (HS) is a rare but life threatening disease caused by inappropriate activation of T-lymphocytes and histiocytes, hypercytokinemia and hemophagocytosis. The most common features are fever, hepatosplenomegaly, unspecified neurological abnormalities, pancytopenia, hyperferretinemia and lipid abnormalities.44,45 This aggressive and potentially lethal disease most often affects infants from birth to 18 months of age but cases in older adults have been reported.46,47 HS was considered as a differential diagnosis for patient number 3, however he did not meet with the revised criteria of the Hstiocyte Society for the diagnosis of HS, 48 there were no changes in bone marrow biopsy and/or cytology compatible wit HS, and moreover, our patient died because of disseminated intravascular coagulopathy. In addition, there are some benign and malignant conditions, including lymphoma and thymoma, that may appear histologically similar to Castleman's disease. Moreover CD may be a precursor lesion of malignancy or concomitant lesion with dendritic follicular sarcoma.26,49,50 Therefore, immunohistologic and immunologic gene rearrangement studies of the specimens can be useful in solidifying the diagnosis. These studies have been used in our patients for sake of accurate diagnosis. Identifying an immunophenotypically varied population of B lymphocytes with polyclonal surface and cytoplasmic immunoglobulin markers helps to confirm the diagnosis of Castleman's disease and differentiate it from lymphoma. 26
Ultrasonography, CT scan, and magnetic resonance imaging have proven to be useful in the diagnosis of masses located in the retroperitoneum. However, the images of CD resemble other masses including lymphoma, tuberculosis, sarcoidosis, and retroperitoneal sarcomas. 51 From radiologic and clinical points of view, disseminated CD may be indistinguishable from lymphoma. Ultimately, biopsy of an enlarged lymph node or mass is mandatory for differential diagnosis. 52
Treatment of localized CD not associated with HHV-8 infection generally is surgical resection, with or without radiotherapy,13,35,53,54 this is almost always curable, resulting in rapid resolution of systemic symptoms and laboratory abnormalities. At present, there is no consensus about the optimal management strategy for patients with the multicenteric form of CD. Successful treatment of such patients has been achieved by using combination chemotherapy with or without prednisone, administered at the time of the initial diagnosis. Many cases, however, are refractory to treatment with corticosteroids or chemotherapy. 27 Treatment with interferon-α or with anti-CD20 monoclonal antibody rituximab (Rituxan; Hoffman-La Roche, Basel, Switzerland) has resulted in durable clinical remission of the disease; however, these findings are based on a small number of patients.55,56 Two of our patients (cases 4 and 8) responded favorably to this drug. Dysregulated overproduction of IL-6 from germinal center B cells is implicated in the pathogenesis of MCD. 33 Previous studies demonstrated that anti-IL-6 receptor antibodies dramatically alleviated the symptoms and biochemical abnormalities, although the disease usually relapsed on cessation of therapy.57,58 Thus, the blockade of IL-6 signaling is considered an attractive approach to treat MCD. However, the long-term efficacy and safety of the humanized anti-human IL-6 receptor antibody need further evaluation.
Twenty-six cases of systemic amyloidosis associated with CD have been reported previously. Seventeen of these had confirmed reactive systemic (AA) amyloidosis (Table 4),59–61 two were confirmed as AL type derived from monoclonal immunoglobulins, and in the remaining seven the amyloid was not characterized.61,62 One of our patients (case-5) with axillary lymphadenopathy had AA amyloidosis complicating MCD. As currently understood, CD is considered to be a heterogenous entity related to conditions of immune deregulation. In this respect, it is interesting that various disorders of the immune system may be characterized by Castleman-like histological changes, such as infections (HIV) and primary autoimmune diseases (systemic lupus erythematosus, POEMS syndrome, etc.60,63 When our patient (case 5) was investigated; hepatitis B, hepatitis C, HIV and other infections were ruled out. The autoantibody pattern and the clinical picture did not point to any primary autoimmune disease. Moreover, the axillary mass, from which CD was subsequently diagnosed, had appeared before many of the organ complications, reinforcing our hypothesis that CD was responsible for triggering the inflammatory response with consequent secondary AA-amyloid deposition which in turn was responsible for the nephrotic syndrome and the orthostatic hypotension. The clinical improvement observed in our patient after the removal of the involved lymph nodes supports this hypothesis. The reversal of both amyloidosis and the nephrotic syndrome, however, are controversial issues, based essentially on case reports, since controlled studies are lacking because of the rarity of the disease. In some reports on monocentric forms, it was shown that the surgical removal of the lymph node mass was curative64–66 and led to regression of the nephrotic syndrome and of amyloidosis. However, a recent report, although confirming the regression of the nephrotic syndrome after excision of an abdominal lymph node, assumed no regression of renal amyloid deposits one year after surgery. 65 Some reports documented no regression of the nephrotic syndrome after lymph node excision,64,66 whereas some showed regression with colchicine therapy.67,68 Treatment of such cases with humanized anti-IL-6 receptor antibodies may lead to improvement of the constitutional and systemic manifestations as well as amyloid regression.69–80
Characteristics of the previously reported patients with AA amyloidosis complicating Castleman's disease.

Summary
we are reporting on a group of patients with a rare disorder, different presentations, and variable outcome. Patients with localized CD who had complete resection of the tumor were cured. Patients with the systemic form who were treated with chemotherapy and rituximab had complete remission.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgment
The authors thank the staff of the Pathology Department at King Fahd Hospital of the University, for their valuable help and support during preparation of this study.