
Abstract
Idiopathic multicentric Castleman disease is a rare and complex disease characterized by systemic inflammation, lymphadenopathy, and multiorgan involvement. This case report presents a 66-year-old Chinese man with idiopathic multicenter Castleman disease without significant lymphadenopathy and challenging diagnosis. Patients present with fever, fatigue, loss of appetite, weight loss, and acute kidney injury. Initially, a urinary tract infection was suspected, but despite anti-infective treatment, the patient’s symptoms persisted. Lymph node biopsy, although there is no significant lymphadenopathy, confirms idiopathic multicenter Castleman disease. Treatment includes thalidomide, cyclophosphamide, and dexamethasone, as well as supportive measures and infection control. After 8 months of follow-up, the patient’s clinical symptoms, inflammatory markers and renal function were significantly improved, and there was no symptomatic recurrence. This case underscores the importance of considering idiopathic multicenter Castleman’s disease in patients with persistent fever and systemic inflammation, even in the absence of significant lymphadenopathy. Early identification and accurate diagnosis of idiopathic multicenter Castleman’s disease can lead to the initiation of targeted therapy strategies that ultimately yield favorable outcomes.
Keywords
Introduction
Castleman’s disease (CD), also known as vascular follicular lymphadenopathy or giant lymphadenopathy, is a rare chronic lymphoproliferative disorder characterized by swollen lymph nodes. 1 It has two main forms: single-center Castleman’s disease (UCD) and multicentric Castleman’s disease (MCD). 2 Patients with MCD who tested negative for human herpesvirus type 8 (HHV-8) were diagnosed with idiopathic MCD (iMCD). 3 UCD is usually mild, usually due to swollen lymph nodes compressing nearby structures. In contrast, iMCD is associated with systemic inflammation and multi-organ involvement, and is characterized by an even distribution of lymph nodes in the neck, axilla, mediastinum, and abdomen, accounting for approximately 40% to 50% of cases. Common clinical manifestations of Crohn’s disease include fatigue (49%), fever (39%), and night sweats (30%). 4 The incidence of Crohn’s disease is estimated to be approximately 21 cases per million person-years, with MCD accounting for about 22% to 24% or 5.1 cases per million person-years. 5 Most cases occur between the ages of 40 and 60 and account for 52% of cases, while less than 10% of cases develop after the age of 65. 5 However, the diagnosis of Crohn’s disease can be challenging, especially in the early stages, as its non-specific symptoms may resemble those of viral infections or autoimmune diseases. In addition, limited awareness of the disease by healthcare professionals can lead to misdiagnosis or delayed diagnosis. Some patients may be incorrectly diagnosed with lymphoma or other cancers, leading to unnecessary treatments that further compromise their health.
In this case report, we describe the clinical course of an elderly Chinese man with Crohn’s disease who initially presents with recurrent fever but lacks significant lymphadenopathy, leading to misdiagnosis. This case highlights the importance of Crohn’s disease as a potential diagnosis in patients with unexplained systemic symptoms or lymphadenopathy, especially when common causes are excluded. It emphasizes the need for health care providers to maintain a high level of suspicion for Crohn’s disease, especially if the presentation is atypical or there is no significant lymphadenopathy. Early diagnosis and timely initiation of appropriate treatment are essential to achieve good outcomes and prevent unnecessary morbidity and mortality associated with Crohn’s disease.
Case presentation
A 66-year-old man with no underlying medical conditions presents with recurrent fever and chills and is referred to hospital for further evaluation. The disease has been going on for 2 months. Laboratory tests at the local hospital showed elevated inflammatory markers, positive white blood cells in the urine, and progressively elevated serum creatinine levels with proteinuria and hematuria. Despite antibiotic therapy, the patient’s symptoms did not improve significantly. After 1 week, fever resolves and serum creatinine levels are low but slightly elevated at discharge.
The patient later came to our hospital with persistent fatigue, loss of appetite and malaise. On admission, the patient’s vital signs were generally within normal limits, but his blood pressure readings were high, blood pressure was 36.6°C, and his oxygen saturation was 98%. The patient appears to be in good spirits, but shows signs of fatigue, slightly pale skin, and pale lips. On lung auscultation, scattered crackles may be heard at the base of the lungs, and there is mild edema in the lower extremities. No splenomegaly or hepatomegaly was found. With no palpable lymph nodes, the patient lost a significant 15 kg of weight over the course of the disease. Further laboratory tests revealed abnormalities in the patient’s renal function and hematologic features. The urine protein level was 3+, and the 24-hour urine protein quantification showed a value of 2.97 g/day. Glomerular hematuria is observed, with a count of 8–10/high power field. The patient had impaired renal function, elevated blood urea nitrogen to 10.74 mmol/L (normal range: 3.6 to 9.5 mmol/L), and serum creatinine level to 128.2 µmol/L (normal range: 57 to 111 micromol/L). In addition, patients had serum albumin as low as 26.2 g/L (normal range: 40 to 55 g/L) and serum globulin as high as 37.5 g/L (normal range: 20 to 30 g/L). White blood cell count 9.36 × 109/L (normal range: 3.5 to 9.5 × 109/L), hemoglobin 96.0 g/L (normal range: 130 to175 g/L), platelet count 250 × 109/L (normal range: 125 to 350 × 109/L). The patient’s hemoglobin level showed a progressive downward trend after admission (Figure 1), indicating the presence of normal pigmented anemia. Reticulocyte testing did not reveal any abnormalities. The patient had a ferritin level of 615.3 ng/mL (normal range: 23.9 to 336.2 ng/mL) and a transferrin-iron binding capacity of 45%. Notably, no symptoms of gastrointestinal bleeding such as gastrointestinal bleeding, melena, or hematemesis were reported. Tumor markers are negative, and associated imaging, bone marrow biopsy, and PET/CT (positron emission tomography/computedtomography) are performed in other hospitals None of the scans showed malignancy as the cause of anemia. A positive direct antiglobulin test indicates the presence of antibodies covering red blood cells. On the 4th day after admission, the patient developed a recurrence of high fever with chills. Laboratory markers associated with infection, including elevated white blood cell count (WBC), interleukin-6 (IL-6), and C-reactive protein (CRP) (Figure 1 shows changes in leukocyte levels, Figure 2 for changes in IL-6 and CRP levels). At the same time, erythrocyte sedimentation rate is 41 mm/hour (normal range: 0.0–15.0 mm/hour) and procalcitonin level is 0.16 nanograms/mL (normal range: <0.05 nanograms/mL). These results indicate the presence of an infectious process. In addition, the patient’s serum creatinine level decreased after antibiotic treatment, suggesting that anti-infective therapy was effective (Figure 3).

Changes in levels of WBC and HGB of the patient during the observational period.

Changes in levels of CRP and IL-6 of the patient during the observational period.

Changes in levels of Scr and ALB of the patient during the observational period.
The patient’s initial chest CT scan upon admission revealed bilateral pneumonia with mild interstitial pulmonary edema and bilateral pleural effusion (Figure 4(a)). Subsequent chest CT scans during the follow-up period showed an obvious improvement in these pulmonary abnormalities (Figure 4(b)). Further analysis of the patient’s urine sample using next-generation sequencing indicated the presence of Candida infection. The patient’s medical history included complaints of frequent urination and discomfort throughout the illness. Although blood cultures conducted during hospitalization in our facility returned negative results, which may be related to the patient’s history of prolonged anti-infection treatment at other hospitals, the diagnosis of a Candida infection was still considered. Consequently, targeted anti-infection treatment was initiated to address both the pneumonia and urinary tract infection. This treatment led to a significant improvement in the patient’s serum creatinine and urine protein levels.

The chest CT scan of the patient at admission (December 6, 2021) showed the presence of bilateral pneumonia with mild interstitial pulmonary edema and bilateral pleural effusion (a). The bilateral ground-glass opacities and pleural effusion had been absorbed after treatment (December 24, 2021) (b).
The results of serum protein electrophoresis showed elevated γ-globulin at 21.6% (normal range: 9%–16%). Serum levels of immunoglobulin A (IgA) were slightly increased at 497 mg/dL (normal range: 82–453 mg/dL), while IgG and IgM levels were within the normal range. Serum immunofixation electrophoresis indicated the presence of positive IgA and lambda light chain. The ratio of serum-free kappa light chain to lambda light chain was 0.26 (normal range: 0.26–1.65). Urine protein electrophoresis showed a negative result. To rule out myeloma as a potential cause, a bone marrow puncture and biopsy were performed. Lymph node ultrasound results showed heterogeneously enlarged lymph node-like nodules in the left neck area, right axilla, and left inguinal area, with the largest measuring 9.5 × 5.0 mm in the left inguinal area. Pathological findings from the left inguinal lymph node biopsy revealed partially preserved lymph node structure, extensive fibrosis in most areas, open lymph sinuses, and partial lymph follicle atrophy. Scattered plasma cells were present in the interfollicular regions, occasionally accompanied by eosinophil infiltration (Figure 5(a)–(c)). Immunohistochemical staining displayed positive cells for CD138, a specific marker of plasma cells (Figure 5(d)), and negative results for HHV8. Subsequently, a pathological examination of the lymph node tissue confirmed the presence of the plasma cell subtype of Castleman’s disease (PC-CD). Considering that elevated blood vascular endothelial growth factor (VEGF) is often observed in iMCD patients, 6 the patient underwent VEGF testing, which revealed elevated VEGF levels of 261.47 pg/mL (normal range: 0–142 pg/mL), further supporting the diagnosis of iMCD.
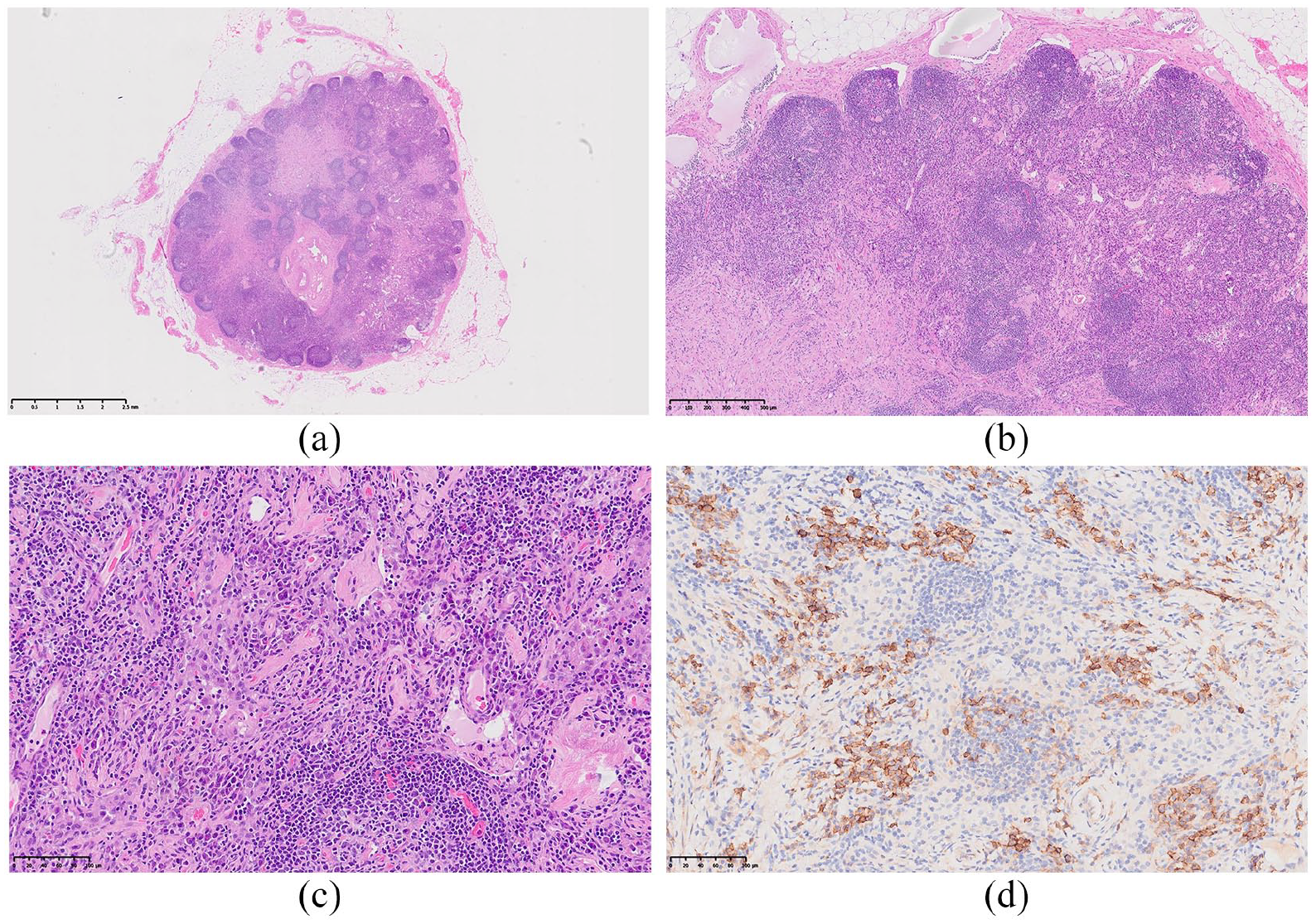
Histological characterization of the left inguinal lymph node of the patient revealed the following findings. (a) The general view of the lymph node. The lymph node structure was partially preserved, but extensive fibrosis was observed in most areas. The lymph sinuses were open, and there was partial lymph follicle atrophy. Scattered plasma cells were present in the interfollicular regions, and occasional infiltration of eosinophils was observed at low magnification (b) and high magnification (c). (d) Immunohistochemical staining of CD138, which is specific to plasma cells.
The patient was prescribed a treatment regimen comprising thalidomide (75 mg/day), cyclophosphamide (150 mg orally three times a week), and dexamethasone (20 mg/week). This therapeutic approach resulted in a significant improvement in the patient’s disease condition. Following discharge, the patient continued the same treatment regimen during the 8-month follow-up period without experiencing any discomfort or adverse effects. Over the course of the follow-up period, several key indicators showed significant improvement. WBC, hemoglobin levels (Figure 1), CRP, and IL-6 levels (Figure 2) demonstrated notable decreases, indicating the resolution of systemic inflammation. Additionally, serum creatinine and serum albumin levels (Figure 3) displayed improvements, signifying stable kidney function and enhanced nutritional status facilitated by anti-infective therapy and nutritional support. The patient achieved complete remission, and these indicators remained stable during the follow-up period, underscoring the effectiveness of the treatment regimen in managing the patient’s condition and maintaining remission.
Discussion
The diagnosis of iMCD can be challenging due to its variable clinical and pathological manifestations, low incidence, and unclear underlying causes. 7 In this reported case, the patient presented with symptoms such as fever, fatigue, poor appetite, and weight loss, which raised suspicion for an underlying condition. A meticulous and comprehensive differential diagnosis was conducted, including the assessment of serum markers such as antinuclear antibody, anti-double stranded DNA antibodies, complement C3, complement C4, and other relevant indicators, all of which returned negative results. Moreover, in conjunction with the patient’s clinical manifestations, conditions like systemic lupus erythematosus, antiphospholipid syndrome, and other autoimmune diseases were ruled out. To confirm the diagnosis of iMCD, a lymph node biopsy was performed. The pathological findings of CD, coupled with the patient’s clinical manifestations and relevant examinations, were meticulously considered following the diagnostic criteria outlined in the 2017 International Evidence-based Consensus Diagnostic Criteria for HHV-8-negative/iMCD. This rigorous diagnostic process helped establish the accurate diagnosis of iMCD in this case. 8
The patient exhibited systemic inflammation characterized by significantly elevated inflammatory markers. Initially, the diagnosis was a urinary tract infection based on the patient’s clinical manifestations, laboratory tests, and imaging findings. However, despite receiving treatment with antibacterial agents, nutritional support, and immunomodulation, the patient’s condition showed fluctuations with changes in body temperature. The levels of IL-6 and CRP in the blood remained consistently high and did not respond well to anti-infective therapy. Although serum albumin levels showed a mild increase after nutritional support, they subsequently declined. While a fever can be a common symptom of various conditions such as urinary tract infection, osteomyelitis, and pulmonary tuberculosis, 9 the unsatisfactory response of the patient’s clinical symptoms and inflammatory markers to treatment raised suspicion for other underlying factors contributing to the observed abnormalities. Patients with iMCD often present with systemic inflammation, including persistent fever and a poor response to treatment targeting infectious microorganisms, which is believed to be associated with cytokine storms. 8 Elevated levels of IL-6 are a characteristic feature of cytokine storms, 10 and research suggests that lymphadenopathy and histopathological changes observed in iMCD are reactive responses to the increased levels of IL-6 and/or other circulating factors during cytokine storms. 3 In this case, the patient exhibited evident systemic inflammation and consistently elevated IL-6 levels, which align with the clinical and laboratory characteristics of iMCD. 11 Studies indicate that IL-6 can induce plasma cell proliferation, elevated CRP levels, VEGF secretion, and thrombocytosis. 6 IL-6 may also contribute to autoimmune phenomena in iMCD by promoting the expansion of CD5-positive B lymphocytes that produce autoantibodies, such as in the case of hemolytic anemia. 12 These mechanisms could potentially explain the observed elevated VEGF levels, positive direct antiglobulin test, and high erythrocyte sedimentation rate in this patient. Hyperimmunoglobulinemia is commonly observed in patients with iMCD, and the possibility of multiple myeloma should be ruled out at the time of diagnosis. 13 In this case, a bone marrow aspiration biopsy was performed to exclude multiple myeloma. This step is crucial to differentiate between iMCD and other conditions and ensure an accurate diagnosis.
The patient initially presented with AKI (acute kidney injury), characterized by a decreased eGFR (estimated glomerular filtration rate) and increased urine protein excretion. The presence of infections, hypovolemia, and a heightened metabolic state suggested that the AKI might have been precipitated by the infection. Treatment primarily focused on symptomatic and supportive measures, including infection control and fluid supplementation, which notably improved the patient’s kidney function and reduced proteinuria. This response further supported the hypothesis that the infection played a role in the AKI. Given the patient’s age, the diagnosis of the PC-CD within the context of iMCD, and the transient and non-severe nature of the kidney function changes, a kidney biopsy was not performed in this case. Nevertheless, existing literature reports indicate that approximately 54% of CD patients concurrently experience kidney damage. 14 In CD, especially iMCD, the primary type of kidney involvement is of the plasma cell or mixed cell subtypes, with a higher prevalence among male patients (84.2%). These patients commonly present with reduced kidney function, often accompanied by proteinuria. Renal biopsies typically reveal thrombotic microangiopathy-like lesions, and chemotherapy has shown potential in reversing kidney damage. 15 Most reported cases indicate that AKI in CD patients can improve with treatment of the underlying disease. However, in this reported case, proteinuria and AKI had already improved before treatment for the underlying CD, suggesting that the transient kidney injury may have been related to the initial infection. It is essential to note that although the patient’s AKI improved with symptomatic and supportive treatments, and the AKI may have been primarily caused by the infection, further clinical observation is necessary to establish a definitive relationship between CD and AKI. Additionally, given that iMCD is the primary type of kidney involvement in CD, clinicians should remain vigilant for kidney damage in CD patients and consider appropriate monitoring and intervention. Notably, thrombocytopenia is a characteristic manifestation of TAFRO syndrome, but the patient’s platelet levels remained within the normal range throughout the disease course. 16 Hence, the diagnosis of TAFRO syndrome in this patient was less likely and was iMCD-NOS 17 (not otherwise specified). During the course of the disease, the patient consistently had low serum albumin levels, especially in the early stages, occasionally dropping below 25 g/L. To mitigate the potential risk of thrombotic complications, clopidogrel was added to the treatment regimen, with discontinuation once serum albumin levels improved.
The pathogenesis of iMCD remains incompletely understood, which has led to a focus on combination chemotherapy as a potential treatment approach. 18 IL-6 has been identified as a key player in the pathogenesis of iMCD, leading to the development of monoclonal antibodies targeting IL-6 as an innovative therapeutic strategy. In the United States, siltuximab stands as the sole FDA-approved drug for iMCD, 19 while tocilizumab is recommended as an alternative to siltuximab in regions where it is not readily available as a first-line therapy. 20 Thalidomide, an immunomodulatory drug primarily used in the treatment of plasma cell malignancies like multiple myeloma, 21 has demonstrated efficacy in managing iMCD by inhibiting various pro-inflammatory cytokines, including IL-6. A phase 2 study involving 25 newly diagnosed iMCD patients who received oral thalidomide, cyclophosphamide, and prednisone showed that 48% of patients achieved symptom relief for at least 24 weeks, meeting the primary endpoint. 22 In this case, tocilizumab was initially recommended as the first-line treatment. However, due to financial constraints and limited drug availability in the patient’s remote area of Urumqi, China, the patient and their family opted for an alternative regimen. Considering the patient’s economic situation and the accessibility of drugs, the treatment regimen of cyclophosphamide, dexamethasone, and thalidomide was chosen. Thalidomide was favored over siltuximab and tocilizumab due to its cost-effectiveness, oral administration, and greater availability. The patient achieved complete remission, with no recurrence during the 8-month follow-up, indicating a positive response to the selected treatment regimen. The patient is continuing with follow-up.
Conclusion
This report details an elderly Chinese patient with iMCD, initially presenting with persistent fever and systemic inflammation but without prominent lymphadenopathy. Diagnosis required a comprehensive assessment of clinical, laboratory, and histopathological findings. iMCD’s rarity and complexity can pose diagnostic challenges, especially for healthcare providers unfamiliar with it, potentially leading to misdiagnosis. To prevent this, healthcare providers must maintain a high suspicion for iMCD in patients with prolonged fever and absent significant lymphadenopathy. Thorough investigations are essential for such suggestive cases. Timely recognition and accurate diagnosis are critical for initiating appropriate treatments and improving patient outcomes. Enhancing awareness of iMCD among healthcare providers is essential for early detection and effective management.
Footnotes
Acknowledgements
The authors would like to thank the patient and his family for their participation in this study.
Author contributions
D.Y., J.Z., H.J., W.L., A.A., and Y.Q. were involved in the clinical data collection and actively participated in the clinical care of the patient. D.L.N. and W.L. drafted the manuscript. H.J. and X.T. conceptualized the research idea and edited the manuscript. J.W. contributed to the revision of the manuscript. C.W. evaluated the lymph node pathology of the patient. All authors contributed to editing and approving the final version of the manuscript.
Availability of data and materials
The original contributions presented in the study are included in the article/supplementary material. For further inquiries, please contact the corresponding author.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Xinjiang Uygur Autonomous Region Project “Research on the key technologies of establishing the operation evaluation model of telemedicine system in autonomous region” (2022A03001-5) to W.L.
Ethics approval
Ethical approval to report this case was obtained from “Clinical Research Ethics Committee of the People’s Hospital of Xinjiang Uygur Autonomous Region (KY2023071701).”
Informed consent
Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
Research registration
Not applicable.