
Abstract
SNP allelic copy number data provides intensity measurements for the two different alleles separately. We present a method that estimates the number of copies of each allele at each SNP position, using a continuous-index hidden Markov model. The method is especially suited for cancer data, since it includes the fraction of normal tissue contamination, often present when studying data from cancer tumors, into the model. The continuous-index structure takes into account the distances between the SNPs, and is thereby appropriate also when SNPs are unequally spaced. In a simulation study we show that the method performs favorably compared to previous methods even with as much as 70% normal contamination. We also provide results from applications to clinical data produced using the Affymetrix genome-wide SNP 6.0 platform.
Introduction
DNA in tumor cells can contain abnormalities in the form of copy number aberrations such as segments with losses or gains of one or several copies of either allele. The lengths of such aberrations can vary between short segments up to an entire chromosome, and their positions are essential both for detecting and for improving knowledge of various sorts of cancer. Therefore, methods that localize copy number aberrations are of great importance. In addition to changes in the total copy number of both alleles together, changes in the
Different techniques to measure DNA copy numbers have been developed, as have methods to evaluate the measurement data. One technique is array comparative genomic hybridization (aCGH), which provides ratios of the copy numbers of a sample DNA, compared to those of some reference DNA. Several different statistical methods have been applied to this kind of data, including different segmentation methods,4,20,21 smoothing6,12 and hidden Markov models.1,7,9,16,19,24,27,28,29 aCGH data provides information only about the total copy number and gives no information about the amount of each allele. Another drawback with such data is the limited number of probes on the arrays. For this reason there is an increased use of single nucleotide polymorphism (SNP) data, which offers denser measurements and provides intensities for the two alleles separately. Using SNP data it is possible not only to estimate copy number changes, but also to find allelic changes such as LOH. Indeed, a copy number amplification may be caused by different allelic changes. For example, a copy number of four could correspond either to {AAAA, AAAB, ABBB, BBBB}, to {AAAA, AABB, BBBB} or to {AAAA, BBBB}, depending on which allele that has gained extra copies.
SNP data has previously been analyzed using various sorts of methods, such as smoothing11,15 and pattern recognition. 22 The most frequently used methods are however based on hidden Markov models (HMMs).3,8,13,17,18,26,30,31 A brief introduction to Markov chains and HMMs is found in Appendix 1. HMMs suit SNP data well since genomic alterations often appear in longer or shorter segments, implying that copy numbers across probes in a small genomic region are correlated. For example, Wang et al 31 and Colella et al 3 model SNP data from the Illumina array, which provides log R ratio data (log2-ratio of total observed intensities to total expected intensities) and BAF data (normalized measure of the relative intensities of the two alleles), using an HMM with six states, while Sun et al 30 apply a more comprehensive model with nine states. Korn et al 13 combine an HMM to model copy number variants with a clustering algorithm to detect genotypes. Li et al 18 also model the proportion of the major allele while Lamy et al 17 use both allelic intensities provided by the Affymetrix array and model them using bivariate Normal distributions.
Several of the methods above assume that the ploidy, ie, mean copy number, of a chromosome is two. This holds for normal cells, but cancer cells are anueploid, ie, their ploidy may differ from two. The necessity for considering ploidy when modeling cancer data is well described by Greenman et al, 8 but in brief one can say that the measured normalized intensity for a probe in a diploid chromosome is twice as large as for a probe with the same copy number in a quadroploid chromosome. Two methods that include ploidy are those of Attiyeh et al 2 and Greenman et al, 8 which both contain a pre-processing step in which the ploidy is estimated. Greenman et al then continue by using an HMM while Attiyeh et al apply a window-based model.
Another feature common in tumor samples, arising from the difficulty to dissect tumor cells only from a tissue sample, is contamination of the tumor cell sample by normal cells. As a result the measured allelic intensities are mixtures of intensities from tumor and normal cells, thus yielding non-integer DNA copy numbers. One way to incorporate such contamination is to model total copy numbers of the mixed sample in a non-parametric way,2,29 but this provides limited information about the copy numbers of the cancer cells. Sun et al 30 estimate the fraction of normal tissue contamination using an empirical method and Colella et al 3 write that it is possible to extend their method to handle contamination, but without being more specific. Li et al 18 show that their method can handle a fraction of normal tissue contamination up to 30%, while Lamy et al 17 report a simulation study with slightly better results. Some tumors however form in a manner such that even with microdisection, a significant proportion of normal cells (say 50% or more) can arise in the sample, and none of the above methods provide results that are satisfactory for such high fractions of normal tissue contamination.
The purpose of the present paper is to devise a method to estimate allelic copy numbers, with ploidy and fraction of normal tissue contamination integrated in the model. Indeed, in all of the above papers, ploidy and/or normal fraction are estimated by adding more or less ad-hoc steps to a model that does not account for these parameters in itself. The model reported here is thus particularly suited for cancer data, for which both of these features are common. By including these parameters in the model they can be estimated alongside the other parameters using all data, rather than adding a pre-processing step or empirical methods using only a small subset of the data. In the simulation study presented below, samples with 30%, 50% and 70% normal contamination are simulated and even for the largest amount of contamination, 97% of the probes are reconstructed to the correct copy number state.
An additional feature of our model is that it is based on a continuous-index Markov chain, which accounts for the fact that the SNP probes are often unevenly spread over the genome. The relevance of a continuous-index model was highlighted by Gupta and Mitra
10
(Section 5.3) for the different but related problem of classifying regions of DNA as nucleosome free regions (NFR) or non-NFR using a two-state HMM. Indeed, they showed that with irregularly spaced probes, a continuous-index model can provide substantially better results than a discrete-index model; 99% vs. 85% or 68% correct classifications in simulations for two different arrangements of the probes. Also the methods by Wang et al,
31
Colella et al
3
and Li et al,
18
who apply discrete-index HMMs to SNP data, aim to take distances between probes into account by letting the Markov transition probabilities depend on these distances in different ways. Common to all of these methods is however that the stipulated transition probabilities violate the Chapman-Kolmogorov equation of Markov chains. That is, letting
The paper is organized as follows. The model is described in in Section 3. Section 4 provides results from a simulation study as well as from an application to clinical data. Concluding remarks are given in Section 5.
Data
The data used in this study are the cancer samples in Greenman et al, 8 produced using the Affymetrix genome-wide SNP 6.0 platform. We applied the algorithm to about 15 different cell line and primary tumor samples, representing various cancer forms including breast, lung and renal cancer. The primary tumor sample PD1753a for which results are reported in Section 4.2 are from a clear cell renal cell carcinoma sample. 32
For probes at SNPs the intensities of the two different alleles are provided, while at other positions only a single total copy number intensity is available. Following Greenman et al, 8 the intensities are normalized by first dividing each measurement by the total intensity of the sample (ie, the sum of all probe intensities over the entire genome), to remove chip-to-chip variation. The mean signals for each allele (or probe at non-SNP positions) are then transformed into a copy number intensity and a genotype intensity that are indicators of total copy number and allelic ratio dosages. The model presented below incorporates intensities for SNP probes only, but is easily extendable to include also probes measuring total copy number only; we elaborate on this further in the Discussion. The cancer data is available from the Cancer Genome Project, subject to a manual transfer agreement, and our Matlab code is available on the WWW. 33
The Model
Basic structure
Let there be
Genotype sets for the different states of the Markov chain, sorted in the order given by the total copy number and copy number of the minor allele.

Genotype sets for the different states of the Markov chain, sorted in the order given by the total copy number and copy number of the minor allele.
For each chromosome
With 16 different states there are 240 different types of jumps and equally many transition rates (per chromosome). It is infeasible to estimate such many rates, and to make the model more parsimonious we assume a large number of them to agree. Specifically we assume, for chromosome
Write

Proportions of probes at which the Markov state was incorrectly reconstructed by the Viterbi algorithm with MAP parameter estimates computed by the EM algorithm. Markov transition rates were λ
Further denote by (

and this model applies for the normal Markov state

note that the variances are taken proportional to the squared means. The probe-specific variance factors
We now carry this model further by assuming that for each probe, the allele intensities follow the mean-variance model given by Eqs. (1)–(2) also for genotypes (
The above model specifies the conditional density of

where
As pointed out in the introduction we include the ploidy

This completes the specification of the basic model. As described above, the parameters
As mentioned above it is often difficult to dissect cancer cells without including any surrounding normal tissue, ie, diploid tissue. Such contamination implies that the measured allelic intensities correspond to a mixture of cancer and normal cells. We denote the fraction of normal tissue in the sample by γ, and consequently the fraction of tumor tissue is 1 - γ. Then for a given probe with, as above, copy numbers
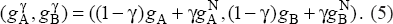
Similarly, the conditional distribution of
Combined genotype sets for the different states of the Markov chain, in a model with normal contamination γ. The weights for the respective combined genotypes are the Hardy-Weinberg weights as in the model without normal tissue contamination, and the total and minor copy numbers for the abberated components are as in Table 1.

The weights for the combined genotypes are Hardy-Weinberg weights as in the model without normal contamination. For example, for a state in Table 2 with three combined genotypes, the weights are
The parameters estimated from a tumor sample are the transition rates λc and η
For a model like the present one, the maximum-likelihood estimator (MLE) typically overestimates the transition rates λ
Finally, to construct an estimate of the trajectory of the hidden Markov chain we use a Viterbi algorithm adapted to continuous-index Markov chains (see Appendix 2.2).
Results
Application to simulated data
To evaluate our method's ability of making correct reconstructions for different amounts of normal contamination, we simulated data from the assumed model, computed MAP parameter estimates using the EM algorithm, reconstructed the hidden Markov chain using the Viterbi algorithm, and finally computed the proportion of probes at which the Markov state was correctly reconstructed. For each simulated dataset we first simulated the Markov chain and the genotypes for each probe position, then computed μ
The simulations were carried out for 30%, 50% and 70% normal contamination, and transition rates λ
To verify the convergence of the EM algorithm we present the EM iterations for three different simulated replicates in Figure 2. The proportions of incorrectly reconstructed probes are plotted in Figure 1.
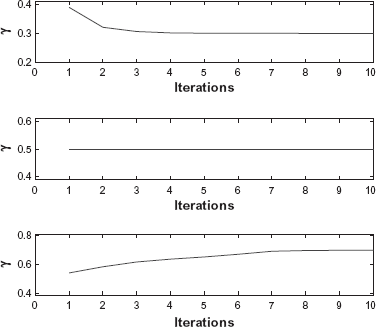
Estimates of normal contamination γ for iterations 1–10 of the EM algorithm and three simulated replicates with different values of γ: γ = 0.3 (top), γ = 0.5 (middle), and γ = 0.7 (bottom). The initial value for γ was 0.5 in all simulations.
These results can be compared to those from the simulation study by Lamy et al. 17 For a normal contamination of 30% the results are similar, but for 45%, which is the largest fraction studied by Lamy et al, their method provides 8%–18% incorrectly estimated probes while at 50% contamination our model provides an error rate below 1%. In addition, the present model performs well even at such a high amount of normal contamination as 70%, when the Markov state is correctly reconstructed at more than 97% of the probes. Obviously the differences between our results and those of Lamy et al depend not only on the different estimation algorithms but also on differences between the number and location of the probes, and on the model for the observed allele intensities and its parameters. However, given the magnitude of the performance improvement, a significant part of it must be attributed to the estimation algorithm as such.
We applied our method to a number of samples from the data described in Section 2. An example is displayed in Figure 3, which shows the Viterbi reconstruction of the Markov chain as well as the corresponding copy numbers compared to the data, for chromosome 3 in primary sample PD1753. For the Gamma priors for λ

Top: Viterbi reconstruction of the Markov path for chromosome 3 in PD1753. Bottom: sum of (standardized) allele intensities for probes within the same chromosome (grey dots), and the copy number of the corresponding state (black solid line).
The reconstruction divides the chromosome into two regions, reconstructed to state 2 ({A, B}) and state 4 ({AA, AB, BB}) respectively. As a simple check of this reconstruction we plotted the standardized allele intensities against each other for all probes in the respective region (Figs. 4–5). Figure 5, corresponding to the normal state, shows three clusters representing the three genotypes AA, AB and BB, while Figure 4 shows four clusters. In Table 1 state 2 is associated to two genotypes, A and B, but with normal contamination this state comprises four combined genotypes (1 + γ)A, AγB, γAB and (1 + γ)B (Table 2). Here γ is estimated at 0.53.

Scatter plot of standardized measured allele intensities in the segment reconstructed to Markov state 2 in Figure 3. The fraction of normal contamination was estimated at 0.53.

Scatter plot of standardized measured allele intensities in the segment reconstructed to Markov state 4 in Figure 3.
For some of the genomes the values of
We have presented a method to estimate the number of copies of each of the two alleles in SNP data, taking three features common in cancer data into account; unequally spaced probes, aneuploidy, and normal contamination. Unequally spaced probes are modeled using a continuous-index Markov chain instead of a discrete-index one, which is the usual choice in the literature. The ploidy and fraction of normal contamination are both included as parameters in the model, which allows us to estimate them along with other variables and using all the data, rather than estimating them separately in a pre-processing step. This set-up also allows us to retain the integer structure of the allele copy numbers. The model's ability to estimate the fraction of normal contamination has been demonstrated in a simulation study, with the results being far better than for previous methods and excellent even with as much as 70% normal contamination.
Above we denoted Markov state 4, ie, the state with genotypes {AA, AB, BB}, the
The emission model, ie, Eqs. (1)–(4), assume that the means and variances of the measured intensities are both linear in the amount of each allele. In practice this assumption may fail, eg, because for large copy numbers the response is nonlinear. One could then include such a non-linearity in the model, and model the mean intensities as
In this paper we have only considered probes that provide allele-specific intensity measurements, but, as mentioned in Section 2, microarrays often also contain probes that measure the total copy number only, ie, the sum of the number of alleles. Such probes can easily be included in our model by speficying a corresponding suitable emission density, ie, a density corresponding to Eq. (3). For instance, this could be a univariate Normal density with mean
Finally we mention some possible limitations of our method. Firstly, the accuracy of the method is likely to be reduced in regions of very high copy number where signal saturation occurs, such as in amplicons, and bespoke nonlinear adjustments may be required (as discused above). Secondly, we have ignored copy number polymorhisms. These will produce non-integer copy numbers in the cancer sample due to the skewed ratio between the cancer and the contaminating normal. If copy number data is available for the normal, it may be possible to generalise these methods to make such an adjustment, however, such regions are generally a lot smaller in scale than the somatic copy number changes seen in cancer and were not considered further. Lastly, we have assumed that the sample in question is derived from a homogeneous collection of cells. However, cell-to-cell variation is quite possibly going to produce a lot of different clones with differing copy numbers, and more general methods will be required to deal with such complexities.
To sum up this paper, copy number variations in cancer are common and their accurate determination is important for determining homozygous deletion, amplifications and breakpoints, all of which can be functionally implicated in cancer. This problem is compounded by normal contamination, making the accurate estimation of integer copy numbers in cancer samples with normal contamination difficult. Here we have introduced a method that addresses this problem.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
Appendix
Acknowledgments
CDG was supported by the Wellcome Trust at the Sanger Centre. The authors would like to thank the anonymous reviewers for their constructive comments and suggestions that improved the presentation of this paper.