
Abstract
Cumulative evidence has shown that structural variations, due to insertions, deletions, and inversions of DNA, may contribute considerably to the development of complex human diseases, such as breast cancer. High-throughput genotyping technologies, such as Affymetrix high density single-nucleotide polymorphism (SNP) arrays, have produced large amounts of genetic data for genome-wide SNP genotype calling and copy number estimation. Meanwhile, there is a great need for accurate and efficient statistical methods to detect copy number variants. In this article, we introduce a hidden-Markov-model (HMM)-based method, referred to as the PICR-CNV, for copy number inference. The proposed method first estimates copy number abundance for each single SNP on a single array based on the raw fluorescence values, and then standardizes the estimated copy number abundance to achieve equal footing among multiple arrays. This method requires no between-array normalization, and thus, maintains data integrity and independence of samples among individual subjects. In addition to our efforts to apply new statistical technology to raw fluorescence values, the HMM has been applied to the standardized copy number abundance in order to reduce experimental noise. Through simulations, we show our refined method is able to infer copy number variants accurately. Application of the proposed method to a breast cancer dataset helps to identify genomic regions significantly associated with the disease.
Keywords
Introduction
Genome-wide association studies (GWAS) are useful for the discovery of genetic variants underlying complex human diseases, such as breast cancer and Type II diabetes.1,2 These genetic association studies typically compare the allele/genotype frequency for each single-nucleotide polymorphism (SNP) between cases and controls. Large projects such as the HapMap and 1,000 Genomes have shown that, in addition to single-nucleotide sequence variations (SNVs), structural alterations, such as copy number variants (CNVs), also account for up to 7.3% of the genetic variation among humans and may be involved in the genetic susceptibility to diseases,3–7 including cancers.8,9
CNVs were first identified in the early 2000s,10,11 and have been found to exist pervasively in human genomes.12,13 Two major platforms of DNA microarrays have been commonly used for copy number estimation, namely, Affymetrix high density SNP arrays and Illumina Bead arrays,14,15 relying on the relative intensity, an indirect measurement of hybridization of fluorescently labeled DNA fragments to immobilized probes on the arrays. Sophisticated statistical models are required to accurately infer the actual copy number within samples. In the past few years, several methods have been proposed for copy number inference. For example, smoothing methods were used in early studies in the field,16,17 which fit a smoothing curve for the intensities along the genomic region and use certain threshold to infer copy number levels. The smoothing methods have been shown to be effective in studies for detecting genomic region with copy number changes. 18 However, these methods suffer from two limitations, namely, difficulty in locating accurate boundaries and difficulty in significance testing for the alterations. 19 Another group of methods adopt certain change-point models for the underlying copy number levels.20,21 A change-point model usually assumes that the SNPs come from segments that are uniformly distributed in human genome, and their underlying copy numbers are piecewise constants with a series of jump points. By maximizing the likelihood function, the parameters as well as the change points can be estimated for copy number inference. Such models are further extended by various formations of hidden Markov models (HMMs).22–25 The HMM assumes that the observed intensities of SNPs are emitted by an underlying Markov chain. It usually explicitly specifies the distribution for the waiting time of copy number changes and the jumping probabilities between copy number states. These methods have emerged as promising tools for copy number inference.
Estimation of array intensity values is challenging due to presence of experimental noise, both within an array and among arrays of different samples. For example, it is commonly known that the level of ozone affects hybridization reactions, which can affect interpretation of the results.26,27 In a recent study, we and others proposed a novel method to estimate copy number abundance on a single-array single-SNP basis, referred to as the probe intensity composition representation (PICR). 28 This method models the cross-hybridization between DNA sequences via their physical binding affinities. It has shown great potential for differentiating copy number signals from background noises. In this article, we propose to extend the PICR method with hidden Markov modeling for copy number inference, referred to as the PICR-CNV. The estimated copy number abundance at each SNP locus from PICR will first be standardized to achieve parity between multiple samples, to which an HMM will be further applied. Our method has two major advantages: 1) By estimating the CNV abundance through PICR, we expect reduction of background noise in intensity values,28–30 and thus be able to boost the performance of HMM. 2) our method does not require between-samples array normalization, which maintains the data integrity and the independency of individual samples. The proposed method is compatible with Affymetrix high density SNP arrays for detection of CNVs.
Methods
This section is organized as follows: We first describe the design of Affymetrix 500K SNP array. Then we briefly review the estimation of copy number abundance for each single array by using a newly established RICR model. 28 We introduce the multi-array standardization of the copy number abundance to achieve equal footing between individuals. Finally, we explain the PICR-CNV by applying an HMM to integrate multiple SNPs for copy number inference.
Design of Affymetrix 500K SNP array
Oligonucleotide microarrays annotate each SNP using a set of 24 probes of 25-mer photolithographically synthesized immobilized nucleic acid sequences. The target sequences are labeled with 3‘-fluorescent dye before hybridization to the array, and their abundance are often measured with the fluorescent intensity on the array after hybridization.31–33 In a 500K SNP array, six quartets are adopted to interrogate a single dimorphic SNP site with its possible alleles commonly denoted as

Distribution of the size of identified CNVs based on BRCA GWAS data.
Estimation for Copy Number Abundance by Picr
The PICR method takes into account the cross-hybridization between DNA sequences via a positional-dependent nearest neighbor (PDNN) model.
28
In PICR, the florescent intensity of a particular probe set is decomposed into four terms: the baseline intensity (

Multi-array Equal Footing by Standardization
By using PICR, the allelic copy number abundance is estimated on a single-array single-SNP basis. Since all the raw fluorescence intensities are subject to experimental scales, which may vary among arrays, it is essential to achieve equal footing for multiple arrays before any further analysis. We propose to define a standardized copy number abundance (SCN) as

where
PICR-CNV: a Hidden Markov Model for Copy Number Inference
Modeling Strategy and Copy Number States
As illustrated by Equation (2), our objective is to detect total copy number changes among subjects. We assume that an interrogated locus covering an SNP may have five possible copy numbers states, with its total copy number ranging from 0 to 4 (Table 1). For simplicity, we also refer to the copy number at an interrogated locus as the copy number of the SNP locus in this article. Such copy number states are not observed directly, and hence, are latent. Following the same notation with existing methods,22,24 the inference of these hidden states is based on two types of observations, log
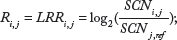
Where

The

where
Configuration of five possible copy number states.

Transition Probability for the Hidden Copy Number States
We assume that the copy number states at SNP loci follow a time-dependent continuous Markov process, with genomic position of SNPs as “time”. The transition probability is dependent on the distance between SNPs. Let

Where

Here,
Emission Probability for the Observations
Since the copy number states are not observed directly, a set of emission probabilities are used to model the distribution of the observed variables (
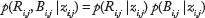
Further, the emission probability of

where

where Φ(.,μ, σ) denotes the cumulative distribution function for a normal distribution with mean μ and variance σ
2
;

Parameter Estimation and Copy Number Inference
In practice, we assume π
Ω = {ω(
μ
σ
μ
σ
The parameters in ω are optimized by using a forward-backward algorithm, also known as the Baum–Welch algorithm. 35 After the parameter estimation, the inference of copy number states is carried out by the Viterbi algorithm. 36 The computational algorithms are commonly used in previous studies, and are not detailed here.
Results
Simulation Study
In the simulation study, we simulated a segment of the genome with length of 10
6
base pairs. We first assumed 10

The parameters for the emission probability of
State: 1 2 3 4 5
μ
σ
The parameters for emission probability of

The observation of
We simulated 100 subjects by using the above model parameters. For each subject, the underlying copy number states and genotypes of 10K SNPs were first simulated in a sequential order according to the transition probabilities. The frequencies of allele
The simulation results are summarized in Table 2. It is seen that the proposed method was accurate for inferring the underlying copy number states when λ
Error rate for inference of copy number states with correctly and incorrectly specified expected length of copy number states.
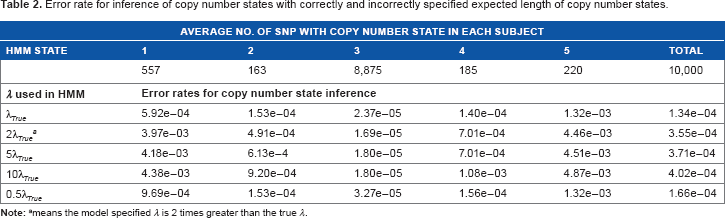
means the model specified λ is 2 times greater than the true λ.
Application to Breast Cancer Data
We also applied the proposed method to study CNVs that are associated with breast cancer development, using a recent GWAS data among a genetically isolated population of Ashkenazi Jews (AJ),
37
in which all participants have their four grandparents of Jewish and of Eastern European ancestry. We are limiting our study to the inherited genetic variation, and potential somatic mutations are beyond the scope of our current study. The original study had three phases. The first phase included 249 breast cancer cases without
We used phase III as an initial study for the analysis. The proposed method was first applied to 10 randomly selected controls for parameter training. The initial genotype calling was conducted by PICR, and all parameters in Ω were optimized and then used to infer the copy number states among all participants. We first examine the distribution of the sizes of identified CNVs (Fig. 1). The shape of this distribution is consistent with existing studies (Fig. 1 of Li et al.
38
). For each SNP locus, we further conducted a Kolmogorov–Smirnov (KS) test to compare the inferred copy numbers between cases and controls. The significant regions were selected if three consecutive SNPs showed significant copy number differences at a level of 1e-07. After the region was selected, a global
Regions showing significant copy number variation in phase III data and their replication in phase I data.

location based on Human Genome Assembly NCBI build 36.1.
Phase III included 243 cases and 187 controls.
Phase I included 249 cases and 299 controls.
We also applied the same procedure to the phase I data for replication. The results are also summarized in Table 3. Among the regions identified from phase III data, the copy number changes remained significant at five regions: 4q31.23, 6q13, 12q23.1, 13q14.3, and 2p21. These five regions contained 10, 5, 4, 4, and 3 SNPs, respectively.
Discussion and Conclusion
In this study, we have proposed an HMM-based method (PICR-CNV) for copy number inference. Through simulations, we have shown that the proposed method is highly accurate for copy number inference and robust against mis-specification of the predetermined model parameter. While it is not straightforward to evaluate the copy number inference with real data due to the unknown copy number status, we have evaluated the proposed standardization approach for genotyping accuracy. We applied PICR to 90 HapMap samples with Affymetrix Mapping 100K arrays, and found that the genotyping accuracies were improved by using standardized copy number abundance compared to using raw copy number abundance (99.70% vs 99.63%). Empirically, we also found that the standardized copy number abundance provided better genotype clustering than its alternative (Fig. 2). The proposed method was further illustrated with an application to breast cancer datasets. The analysis of breast cancer data also identified a few genomic regions that were significantly associated with breast cancer development. Most of these identified regions have been reported in the literature for potential involvement in breast cancer. One SNP in the region 4q31.23 has been recently reported to be significantly associated with breast cancer progression.
39
A gene
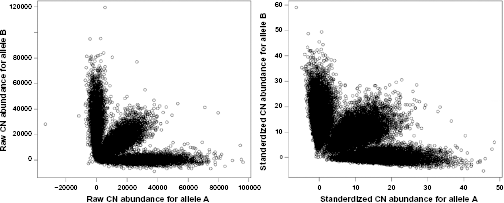
Raw and standardized copy number abundance for a randomly selected HapMap sample (NA12892).
The associations of the identified regions, including 4q31.23, 12q23.1, 13q14.3, and 2p21, were also replicated by using an independent dataset. The region of 4q31.23 was identified by phase III as the one with the largest number of significant SNPs. The long arm of chromosome 6 was reported to be frequently rearranged in human cancers.48–50 The region of 6q13 was among the important regions that showed copy number alterations.51,52 For region 12q23.1, a gene
We are also aware that our method may have a few limitations. First, our copy number estimation method is based on the design of Affymetrix 500K SNP arrays. Further extension will be needed before applying it to Illumina platform or Affymetrix 6.0 arrays. Our current study is a secondary analysis of an existing GWAS dataset, extending previous genotype-based association study to copy-number-based association study. The Affymetrix SNPs array has been a major platform for SNP genotyping and copy number estimation. It was adopted by the Wellcome Trust Case Control Consortium (WTCCC) for intensive GWAS of 14,000 cases of seven common diseases and 3,000 shared controls. 62 Second, the current study only considered the total copy number changes at each locus. However, copy number changes may still occur without total number changes, such as balanced copy number with preferential loss of heterozygosity (LOH). Further extensions are needed to account for such copy number changes. Third, our method currently focuses on detecting the total copy number changes in an unrelated population. Detecting copy number status for related individuals or paternal (maternal) specific copy numbers is beyond the scope of current study.
Author Contributions
Conceived and designed the experiments: ML, WF. Analyzed the data: ML, YW, WF. Wrote the first draft of the manuscript: ML, WF. Contributed to the writing of the manuscript: YW. Agree with manuscript results and conclusions: ML, YW, WF. Jointly developed the structure and arguments for the paper: ML, WF. Made critical revisions and approved final version: YW, WF. All authors reviewed and approved of the final manuscript
Footnotes
Acknowledgments
We would like to thank Dr. Burt Gold from the National Cancer Institute and Drs. Vijai Joseph and Kenneth Offit from the Memorial Sloan Kettering Cancer Center, for helping us access the BRCA dataset and also for their critical comments that improved this manuscript. This work was supported in part by the Michigan State University College of Medicine Summer Oncology Research Scholarship and the University of Arkansas for Medical Sciences College of Medicine Children's University Medical Group Fund Grant Program.