
Abstract
Survival analysis in biomedical sciences is generally performed by correlating the levels of cellular components with patients’ clinical features as a common practice in prognostic biomarker discovery. While the common and primary focus of such analysis in cancer genomics so far has been to identify the potential prognostic genes, alternative splicing – a posttranscriptional regulatory mechanism that affects the functional form of a protein due to inclusion or exclusion of individual exons giving rise to alternative protein products, has increasingly gained attention due to the prevalence of splicing aberrations in cancer transcriptomes. Hence, uncovering the potential prognostic exons can not only help in rationally designing exon-specific therapeutics but also increase specificity toward more personalized treatment options. To address this gap and to provide a platform for rational identification of prognostic exons from cancer transcriptomes, we developed ExSurv (https://exsurv.soic.iupui.edu), a web-based platform for predicting the survival contribution of all annotated exons in the human genome using RNA sequencing-based expression profiles for cancer samples from four cancer types available from The Cancer Genome Atlas. ExSurv enables users to search for a gene of interest and shows survival probabilities for all the exons associated with a gene and found to be significant at the chosen threshold. ExSurv also includes raw expression values across the cancer cohort as well as the survival plots for prognostic exons. Our analysis of the resulting prognostic exons across four cancer types revealed that most of the survival-associated exons are unique to a cancer type with few processes such as cell adhesion, carboxylic, fatty acid metabolism, and regulation of T-cell signaling common across cancer types, possibly suggesting significant differences in the posttranscriptional regulatory pathways contributing to prognosis.
Introduction
Survival analysis is a statistical approach to evaluate an event or study where the outcome is based on the vital status of the samples. 1 Cancer treatment and clinical trials are two common use cases for analyzing the survival of the samples based on the given treatment. However, over the recent years, several major web servers that calculate the survival contribution of mRNAs by employing publicly available clinically annotated breast cancer microarray data have become available including Kaplan–Meier plotter, 2 GOBO, 3 RecurrenceOnline, 4 and bc-GenExMiner. 5 PrognoScan 6 is one of the first studies integrating microarray expression data from Gene Expression Omnibus (GEO) for multiple cancer types to predict the prognostic mRNAs. As a result of increase in the availability of sequencing-based genomic data along with clinical annotations across cancer types from various consortiums such as The Cancer Genome Atlas (TCGA)7–9 (http://cancergenome.nih.gov/) and the International Cancer Genome Consortium (ICGC) (https://icgc.org/), cancer prognostic biomarker identification and comparison among them has become the major outcomes of various multi-omic studies. SurvExpress 10 and PROGgene 11 have integrated multiple RNA sequencing (RNA-seq) expression profiles from GEO datasets and report the contribution of an mRNA expression as a prognostic biomarker. SurvNet 12 is an online service for identifying the network-based biomarkers associated with clinical information. It is preloaded with TCGA data and reports the survival significance in mRNA and protein levels. cBioPortal 13 is a comprehensive web-based platform that integrates several cancer genomic datasets including those from TCGA. However, all of the listed services only report the prognostic properties and survival contribution at mRNA level. Alternative splicing is a posttranscriptional gene regulatory mechanism that contributes to different protein products due to alterations in the combination of exons originating from the same gene. 14 Exon inclusion/skipping is a class of a splicing event, where in an exon is included or missed in the final isoform product. 14 Hence, identifying the potential prognostically involved/missed exons in cancer genomes would be beneficial for a better understanding of not only the involvement of posttranscriptional regulatory alterations in cancers but would also provide novel insights into potential exon-level functions in tumor evolution. For example, SRSF2, an RNA-binding protein, mutations drive recurrent mis-splicing of key hematopoietic regulators such as EZH2 in patients with myelodysplastic syndromes. 15 Similarly, mutant U2AF1, an RNA-binding factor, alters downstream gene isoform expression by altering the splicing mechanism. 15 A recent study 16 analyzed genome-wide patterns of RNA splicing across 805 matched tumor and normal control samples from 16 distinct cancer types to identify signals of abnormal cancer-associated splicing. This study reports intron retention, a category of alternative splicing, to be common across cancers even in the absence of mutations directly affecting the RNA splicing machinery. In light of several recent studies providing support for extensive rewiring of posttranscriptional regulatory networks in multiple cancer types,16–19 there is an immediate need for resources that can enable rapid mining and validation of high-confident prognostic alternative splicing events in cancer transcriptomes.
In this study, we present ExSurv (https://exsurv.soic.iupui.edu/), which to our knowledge is the first online web server that provides exon-level survival significance by using the RNA-seq expression datasets and the associated clinical metadata for four cancer types from the TCGA project7,9 (http://cancergenome.nih.gov/). We precalculated the prognostic significance of more than 600,000 annotated exons in Ensembl
20
using survival package
21
in R across the four cancer types. We stored the TCGA clinical data, exon survival
Materials and Methods
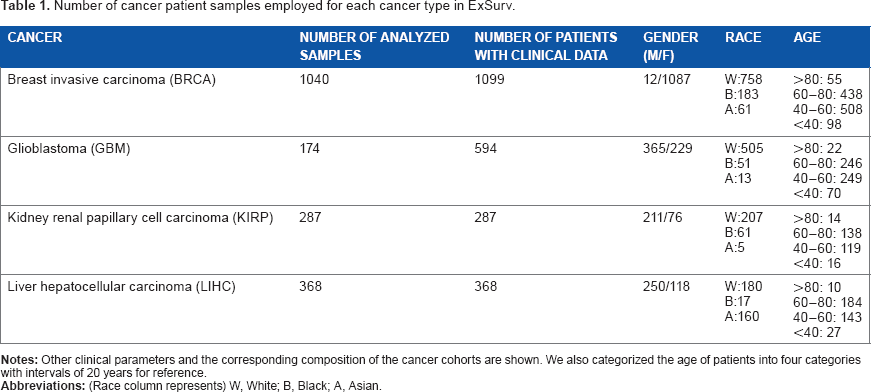
Cancer Types and the Corresponding Number of Cancer Samples Analyzed in this Study from the TCGA Project
We selected four cancer types, namely, breast invasive carcinoma (BRCA), liver hepatocellular carcinoma (LIHC), glioblastoma (GBM), and kidney renal papillary cell carcinoma (KIRP), and quantified the exon expression levels for patients with accessible raw RNA-seq data. It is important to note that TCGA is a collaborative effort; hence, the generated data could be from different dates and platforms. However, it is not suggested to do batch effect correction since it may bring additional noises to the analysis. The majority of TCGA cancer patients are
Number of cancer patient samples employed for each cancer type in ExSurv.
Workflow for Processing RNA-seq Data and Quantification of Exonic Expression Levels
Hierarchical indexing for spliced alignment of transcripts (HISAT)
22
is a highly efficient alignment tool for aligning short reads from RNA-seq experiments onto reference genome. HISAT claims to be the fastest alignment system currently available, with better accuracy than most other methods. We ran HISAT with – dta parameter options to align each of the RNA-seq samples to generate corresponding Binary Alignment/Map (BAM) files using the human genome reference 38 annotations obtained from Ensembl.
23
We then quantified the expression of exons using StringTie,
24
a computational method that applies a network flow algorithm together with optional de novo assembly, and estimated the multimap corrected number of reads for every annotated exon in human genome build 38. To normalize the number of reads mapped to genomic regions, transcript per million is proposed by Li and Dewey,
25
which corresponds to normalized read count value based on the length of the transcript isoform under consideration and the total number of mapped reads in the whole genome. We employed a similar approach to quantify the exon expression, normalized by the length of exon and the total number of the reads mapped to the whole genome. Here,

Clinical Metadata from TCGA Samples Employed for Survival Analysis in ExSurv
Clinical data are crucial for survival analysis. TCGA (http://cancergenome.nih.gov/) provides both sequencing data and the corresponding clinical information for most of the patients for each cancer type included in ExSurv. The clinical data included the vital status of a patient, number of days from the first day of medication till the last follow-up, gender, cancer subtypes, and other cancer-specific information. We included vital status and the number of days since last follow-up in our survival analysis.
Survival Analysis for Exons to Identify Prognostic Markers in a Cancer Type
We employed survival package in R26,27 to estimate the contribution of a given exon to survival of patients in a cancer type based on their expression. We divided the cancer patients based on the expression of a given exon into two groups (ie, High and Low). The group that was labeled as
Construction of the Database Backend for Serving the User Interface of ExSurv
We aligned the raw RNA-seq datasets from the cancer samples against HG38 and used the exon annotations from Ensembl database, 23 as discussed above. The exon survival information and metadata associated with cancer patients were organized into separated tables. Likewise, the genomic annotations were organized into separate tables (Supplementary Fig. 1 for a schema of the ExSurv database). The design of the database enables updating either the annotations or precomputed datasets or both without extensive dependencies, thereby speeding up updates to the ExSurv when there are changes in the datasets without the need for a structural change in the database. We stored the data required for our service in a MySQL database. We also developed PHP scripts to handle user queries and generate exon-level survival plots by integrating preprocessed data from the database to perform dynamic survival analysis using the queried data in R with survival package. 26
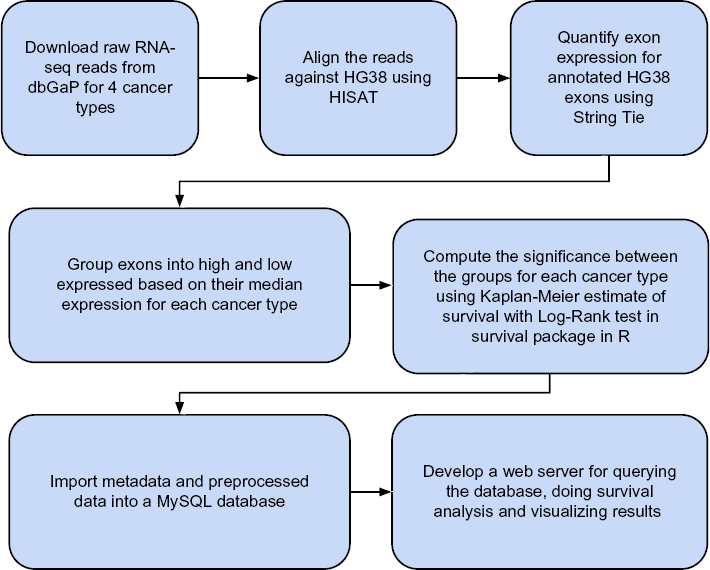
Major steps involved in the generation of exon-level survival estimates in the ExSurv database and visualization engine. As outlined in the flowchart, our analysis pipeline comprises obtaining the raw RNA-seq datasets for more than 1800 patients spanning four different cancer types along with their clinical metadata. Raw RNA-seq was processed using HISAT
22
aligner to generate BAM formatted files, and StringTie
24
was used for exon-level expression quantification. Exon expression level across patients in a given cancer type along with clinical information on the follow-up time periods were used to generate Kaplan–Meier plots and corresponding
Framework Employed for Building the User Interface of ExSurv
We implemented a simple interface for users to search for their gene of interest and visualize the exon-level survival analysis results in the selected cancer type. We use JavaScript integrated with PHP scripts to query the database and retrieve the expression results that are then provided as input for survival analysis in R. ExSurv provides the users to not only search for genes of interest and naturally all the exons associated with it but to also limit to certain level of significance. The results of the search are listed as survival plots for each exon corresponding to the gene that matches the query (eg, a gene symbol or Ensembl gene ID). The metadata related to that exon in the context of the cancer samples analyzed is also given next to its survival plot. The users can export the exon-level survival plots for storing them locally. We also implemented an export functionality for downloading raw expression data as well as the associated metadata across cancer samples analyzed for each exon. ExSurv does not require users to log in, and it is freely available on https://exsurv.soic.iupui.edu/.
Results and Discussion
Overview of Datasets and Approach Employed for Building ExSurv
Figure 1 shows the major steps involved in the generation of exon-level survival estimates as implemented in ExSurv database and visualization engine. Briefly, we obtained the raw RNA-seq datasets for individual patients from CGHub portal (https://cghub.ucsc.edu/) after an approval process from the dbGaP (http://dbgap.ncbi.nlm.nih.gov/) to access the raw datasets for various cancer types. Grouping of the patients based on exon levels together with the vital information of the patients and the time-to-event information for each cancer type were used for survival modeling by employing the Kaplan–Meier estimate with log-rank test (see “Materials and methods” section). Resulting datasets were stored in a MySQL database as described below and a visualization engine built for dynamic access to the underlying data.
Four tables were designed to store the gene–transcript–exon relationship (Supplementary Fig. 1). “Gene” table has Ensembl gene IDs, their corresponding symbols, and gene synonyms to facilitate searches when users query for alternate gene names in addition to those reported as standard HUGO Gene Nomenclature Committee (HGNC) gene symbols (http://www.gene-names.org/). “Transcript” and “Transcript_Exon” tables show the transcript IDs associated with each gene and exons that are associated with each transcript, respectively. Since exons are the building blocks of ExSurv, their genomic coordinates, associated chromosomes, and strand information are organized in the “Exon_Info” table that is connected with the “Transcript_ Exon” table. “Cancer_Patient_Info” table stores the clinical information retrieved from TCGA portal such as vital status, number of days from the first day of medication till the last follow-up, and gender for all the patients analyzed in this study for each cancer type. Table 1 shows the composition and the number of patients with various clinical attributes for the four cancer cohorts employed in this study. We stored the precalculated survival probability of each exon across every cancer type in “Cancer_Exon_Survival” table. “Exon_Survival_Data” contains the generated expression data for each exon along with its corresponding expression group, needed for survival analysis and for generating the survival plots (Supplementary Fig. 1 for the complete database schema).
ExSurv Provides a Rapid Means of Visualizing the Exon-Level Survival Contributions for All Exons in a Given Gene
ExSurv has a user friendly web interface for querying and visualizing the exon-level survival contributions for all exons associated with a given gene symbol or Ensembl gene ID. It has a search form placed next to its main navigation bar to make it accessible from everywhere, and the form includes

Screenshots from ExSurv user interface. (
Most Prognostic Exons are Cancer Specific
We extracted the high-confident prognostic exon biomarkers across each of the cancer types studied here at a survival probability <0.01 to study their overlap across cancer types. A comparison of the prognostic exons revealed that while more than 40,000 exons were significant for BRCA, only 4000–7000 significant exons were found for other cancer types. This can be due to a number of reasons including the variability in the sample sizes such as the higher coverage for BRCA compared to the other cancer types and heterogeneity in the cancer samples for all cancer types may not be the same. Nevertheless, a comparison of the number of exons, which were found to be significant across two or more cancer types, clearly revealed a surprising trend (Fig. 3). We calculated the pairwise significance of the overlap among the cancers using hypergeometric test and found that the reported overlap values between all the pairs are indeed significant (
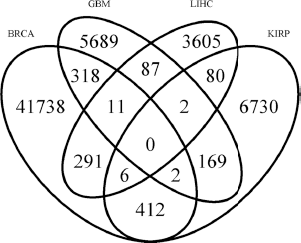
Venn diagram showing the number of prognostic exons in four different cancer types. Significant majority of the exons contributing to patients’ survival were found to be cancer specific. Only exons that were found to be significant at a
Functional Analysis of Genes Associated with Survival-Associated Exons Reveals Unique as Well Core Functional Signatures of Cancer Types
Performing a functional analysis of the prognostic exons against the current literature is not yet possible as very few studies have attempted to analyze the exon-level contributions to cell growth phenotypes. However, it is possible to analyze the genes associated with the prognostic exons, and hence, we extracted the list of top 500 genes with highest number of prognostic exons, contributing to patients’ survival in each cancer type to understand the biological processes associated with such exons. Figure 4 shows the analysis of the enriched biological processes associated with the extracted genes for each cancer type using ClueGO
28
against Gene Ontology database
29
with a
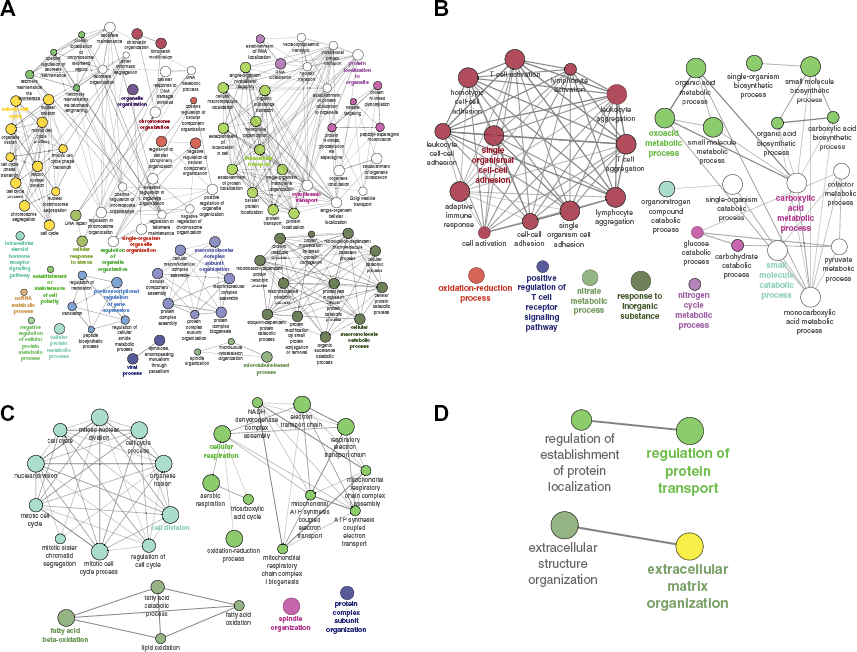
Functional enrichment using ClueGO for genes associated with exons found to be significant in (
Conclusions
Cancer is a complex multifactorial disease with our understanding of the posttranscriptional mechanisms contributing to or causal to the cancer phenotypes being very limited. Most approaches currently focus on identifying prognostic markers at the individual gene or transcript level within a cancer type or across cancer types; however, our understanding of the post-transcriptional mechanisms altered in cancer transcriptomes or the resulting splicing biomarkers are limited. Hence, identifying and understanding the function of cancer type-specific prognostic biomarkers based on such poorly characterized layers of regulation is challenging and has recently gained lot of attention in precision medicine field. Alternative splicing is a posttranscriptional mechanism that might change the expression level of the resulting mRNA isoform and hence the final protein product consequently, thereby causing aberrations in the downstream interactome and disease phenotypes. In particular, exon inclusion/exclusion is one of the well-studied alternative splice forms that results in different protein products. While a complete and comprehensive understanding of the functions of exons in the context of their corresponding functional transcripts is still premature, with the availability of novel CRISPR/Cas9 genome editing screens, it might be increasingly possible to study the impact of individual exons on cancer phenotypes in order to rationally design exon-specific therapeutics to decrease the of-target effects and to increase specificity towards more personalized treatment options for cancers. To address this gap and to provide a platform for rational identification of prognostic exons, in this study, we developed ExSurv, a database and web server for exon-level survival significance for four cancer types. ExSurv is a web-based platform where a user can query a gene of interest in a cancer type, resulting in the expression levels of all the exons associated with the gene along with their survival significance and associated plots to be visualized online or for local use.
Our analysis of the resulting prognostic exons across four cancer types clearly revealed that most of the survival-associated exons are unique to a cancer type with few associated processes common across cancer types, possibly suggesting significant differences in the posttranscriptional regulatory pathways contributing to prognosis. ExSurv is a fully functional proof-of-concept platform that will be improved by adding additional cancer types and other functionalities in future versions. Current version of ExSurv performs the survival analysis of exons among the patients of a selected cancer type without employing additional clinical features such as gender and subtype. We will also improve the current platform to facilitate exon survival analysis only among patients who match specific clinical features. Currently, such filtering frequently limits the number of samples, thereby decreasing the power of the analysis for most cancer types. In addition, RNA-seq data for most of the cancer types from TCGA project have been sequenced from multiple sequencing centers or platforms, which could potentially add batch effects to the downstream analysis. However, since such postprocessing and stratification can further reduce the power of the data, careful considerations should be included in the pipelines. This becomes especially important as the number, source, and heterogeneity of the samples increase due to contributions from initiatives such as the ICGC. 31 We anticipate that future versions of ExSurv, which can accommodate such stratifications and controls, could serve to become very powerful for studying the impact of prognostic exons even in subtypes of cancers.
Author Contributions
Conceived and designed the study: SCJ, SH. Downloaded, preprocessed the datasets, and imported the preprocessed data into the database: SH. Developed the web server: GB. All the authors contributed to the generation of the material needed for writing the manuscript. All the authors reviewed and approved the final version of the manuscript.
Footnotes
Acknowledgment
The authors wish to thank the members of the Janga lab for helpful discussions in the course of the study.