
Abstract
Tumor suppressor gene, STK11, encodes for serine-threonine kinase, which has a critical role in regulating cell growth and apoptosis. Mutations of the same lead to the inactivation of STK11, which eventually causes different types of cancer. In this study, we focused on identifying those driver mutations through analyzing structural variations of mutants, viz., D194N, E199K, L160P, and Y49D. Native and the mutants were analyzed to determine their geometrical deviations such as root-mean-square deviation, root-mean-square fluctuation, radius of gyration, potential energy, and solvent-accessible surface area using conformational sampling technique. Additionally, the global minimized structure of native and mutants was further analyzed to compute their intramolecular interactions and distribution of secondary structure. Subsequently, simulated thermal denaturation and docking studies were performed to determine their structural variations, which in turn alter the formation of active complex that comprises STK11, STRAD, and MO25. The deleterious effect of the mutants would result in a comparative loss of enzyme function due to variations in their binding energy pertaining to spatial conformation and flexibility. Hence, the structural variations in binding energy exhibited by the mutants, viz., D194N, E199K, L160P, and Y49D, to that of the native, consequently lead to pathogenesis.
Introduction
Cancer is driven mostly by somatic mutations. Efficiently identifying the driver mutations from a vast majority of passenger mutations, which do not contribute to cancer, is a difficult task. The gene STK11 is a tumor suppressor gene and encodes for serine-threonine kinase, which has a critical role in regulating cell growth and apoptosis.1,2 Inactivation of this gene leads to development of cancer. Many mutations in STK11 are small deletions or point/missense mutations that are present in the STK11 catalytic kinase domain, and a few of them occur within the noncatalytic COOH- terminal, thereby resulting in STK11 protein reduction, loss, or inactivation.3,4 Mutations in the STK11 gene also cause Peutz-Jeghers syndrome, a condition characterized by the development of noncancerous growths called hamartomatous polyps in the gastrointestinal tract. 5
The catalytic kinase domain (residues 43–347) of STK11 associates with the pseudokinase STe20-related adaptor (STRAD; residues 59–431) and full-length scaffolding mouse protein 25 (MO25) in a 1:1:1 heterotrimeric complex in the cell.6–10 In contrast to the majority of the protein kinases that are regulated by phosphorylation, STK11 is activated by binding to STRAD and MO25 through an unknown, phosphorylation-independent molecular mechanism. Although STK11 lacks phosphorylation of the activation loop, it adopts an active conformation. The αC-helix of STK11 is rotated into the canonical closed conformation, by forming the conserved salt bridge between Lys (78) and Glu (98). This active conformation of STK11 appears to be achieved through contributions of both STRAD and MO25. The C-terminal lobe of STRAD interacts with both N- and C-terminal lobes of STK11 kinase domain. Mutations in STK11 can lead to its inactivation without affecting this complex assembly. 6
Compared to our previous study, 10 we have suggested a distinct computational approach to analyze the functional impacts of selected mutations of STK11 in pathogenesis. Molecular dynamics simulation protocol and thermal annealing process were used to compare the native and mutants, viz., D194N, E199K, L160P, and Y49D. Mutant D194N has been reported in lung cancer 11 ; E199K, reported in large intestine cancer 12 ; L160P, reported in cervical cancer 13 ; and Y49D, reported in skin cancer. 14 The computational method followed here might distinguish the driver mutations of cancerous genes from a vast number of passenger mutations.
Materials and Methods
Datasets
The protein sequence and variants of STK11 were obtained from the Swiss-Prot database15,16 available at http://www.expasy.ch/sprot. The 3D Cartesian coordinates of the protein STK11 were obtained from Protein Data Bank (PDB Id: 2 WTK) for in silico mutation modeling and docking studies. 17
Modeling Missense Mutation on Protein Structures and Energy Minimization
SWISSPDB viewer 18 was used for performing mutant modeling on STK11, and NOMAD-Ref server was used for performing the energy minimization for 3D structures. 19 GROMACS force field embedded in NOMAD-Ref was used for energy minimization, based on the steepest descent, conjugate gradient, and limited-memory Broyden-Fletcher-Goldfarb-Shanno methods. It creates a GROMACS topology using the GROMOS96 vacuum force field. 20
Prediction of Disease-Causing Mutations by Artificial Neural Network Predictor, NetDiseaseSNP, and Validation by Catalog of Somatic Mutations in Cancer Database
For the prediction of disease-causing mutations, we used the tool NetDiseaseSNP, 21 a sequence conservation-based predictor of the pathogenicity of mutations, which exploits the predictive power of artificial neural networks. This method derives sequence conservation from position-specific scoring matix (PSSM), based on the alignment algorithm of sorting intolerant from tolerant (SIFT), which is complemented with the calculation of surface accessibility by the predictor NetSurfP. 22 This approach provides NetDiseaseSNP the potential to extract all relevant information directly from protein sequences. NetDiseaseSNP encodes the SIFT score (normalized probability) for the SNP amino acid in one input neuron. SIFT predicts the effects of all possible substitutions at each position in the protein sequence. This server is available at http://www.cbs.dtu.dk/services/NetDiseaseSNP. The artificial neural networks of this predictor will generate an output value close to 1 if the combination of features describing that particular mutation suggests that it might be involved in disease, and close to 0 for neutral mutations.
The database Catalogue of Somatic Mutations in Cancer (COSMIC) 23 is the largest and ample resource for exploring the impact of somatic mutations in human cancer. In order to gain a deep sense of knowledge on the key cancer genes, many appropriate literatures were identified for each gene and then subjected to manual curation. This manual curation allows this database to capture very high detail across mutation positions and disease descriptions. The variants were subjected to a COSMIC search to extract the information of primary tissue affected. The COSMIC dataset can be assumed to be enriched for cancer driver mutations when compared with large-scale somatic mutation discovery datasets, which were expected to contain a fair number of passenger mutations. 23
Ensemble Analyses through Normal Mode-Based Simulation
Conformation sampling approach was used to generate ensembles to expand the chances of identifying an energetic landscape that closely matched the input structures. 24 The Normal Mode-based Simulation (NMSim) approach 25 has been shown to be a computationally efficient alternative to molecular dynamics simulations for conformational sampling of proteins and performs three types of simulations, viz., unbiased exploration of conformational space, pathway generation by a targeted simulation, and radius of gyration (RoG)-guided simulation. This Web server implements a three-step approach for multiscale modeling of protein conformational changes. Initially, the protein structure is coarse-grained, followed by a rigid cluster normal mode analysis that provides low-frequency normal modes, and finally, these modes are used to extend the recently introduced idea of constrained geometric simulations by biasing backbone motions of the protein, whereas, side chain motions are biased toward favorable rotamer states (NMSim). This program is accessible through http://www.nmsim.de. The RoG-guided simulation type was used here to generate the ensembles of native and mutant structures. The parameters used for the rigid cluster decomposition were as follows: energy cutoff for hydrogen bonds (-1.0 kcal/mol), method for placing hydrophobic constraints (3), cutoff for including hydrophobic constraints (0.35 A). The method chosen for the normal mode analysis was rigid cluster normal mode analysis, and the distance cutoff for interactions between C-alpha atoms was set to 10 A. The parameters for the simulation were as follows: number of trajectories (1), number of simulation cycles (500), number of NMSim cycles (1), frequency of writing out conformations (1), side chain distortions (0.3), normal mode range (1–50), ROG mode (1), and step size (0.5 A). The structural diversity of native and mutant ensembles, obtained through NMSim program, was evaluated by their root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF). Geometrical and conformational deviations of native and mutant ensembles were studied through RMSD. 26 RMSF was used to study the fluctuation of residues present in native and mutant structures, which represents their atomic mobility.27,28 RoG is indicative of the level of compaction in the structure 29 and calculated using VEGA ZZ package, 30 which implements trajectory visualization and numerous calculations. Packing defects and folding patterns of native and mutants were studied through solvent-accessible surface area (SASA), 31 which was computed using VEGA ZZ package. 30 Changes in SASA are caused due to changes in their tertiary structures, which in turn are affected by the folding patterns of native and mutants. 32
Furthermore, native and mutant ensembles were analyzed for their energetic contributions through Bayesian Analysis Conformation Hunt (BACH) algorithm (http://bachserver.pd.infn.it/in) in which the all-atom energy score was computed based on 1091 parameters. The BACH score was also used to discriminate the global minima of native and mutant ensemble.33,34
Analysis of Protein Integrity by Computing Interactions, Hydrogen Bond Analysis, Simulated Thermal Denaturation, and Secondary Structure Analysis
Protein stability relies on various strong and weak interactions. 35 Protein interactions calculator (PIC) program, 36 a structure-based algorithm was used to compute the interactions stabilizing native and mutant structures. The program was used to compute the disulfide bridges, interactions involving hydrophobic, ionic, aromatic–aromatic residues, aromatic–sulfur residues, and cation-π interactions, which contributed to the overall integrity of native and mutant structures. In addition, the hydrogen bond analysis was done, as they stabilize secondary structure elements. 37 HB Plot (http://virtuadrug.com/products/hb-plot/index.html) is a tool for exploring the protein structure and function by describing the structure as a network of hydrogen bonding interaction. This Web server plots three classes of hydrogen bonding by color coding: type 1 - short (distance smaller than 2.5 Å between donor and acceptor), type 2 - intermediate (between 2.5 Å and 3.2 Å), and type 3 - long hydrogen bonds (greater than 3.2 Å).37,38 Besides, the native and mutant structures were subjected to simulated thermal denaturation through Proflex, embedded in StoneHinge program available at http://stonehinge.bmb.msu.edu to describe the folding patterns. 39 This Web server simulated incremental thermal denaturation of the structure, as the calculated temperature rises, and hydrogen bonds weaker than the current energy level were broken. Hydrophobic interactions were maintained throughout the process, as the strength of these interactions increased somewhat with a modest increase in temperature. 40 Furthermore, secondary structure elements were analyzed through STRIDE program, which assigns the secondary structure based on combined use of hydrogen bond energy and statistically derived backbone torsional angle information. 41 Changes in α helix, β sheets, and coils of native and mutants were compared and visualized, as structural deterioration might cost the functional properties of structures.
Analysis of Interaction of Native and Mutant with STRAD
We subjected the native and mutants for docking studies by using PatchDock42,43 in order to understand their functional activity with the binding partner, STRAD. PatchDock is a geometry-based molecular docking algorithm, and it is aimed at finding docking transformations that yield good molecular shape complementarity. 42 Such transformations, when applied, induce both wide interface areas and small amounts of steric clashes. A wide interface ensured that several matched local features of the docked molecules that have complementary characteristics were included. The PatchDock algorithm divides the Connolly dot surface representation43,44 of the molecules into concave, convex, and flat patches. Then, complementary patches were matched to generate candidate transformations. Each candidate transformation was further evaluated by a scoring function that considered both geometric fit and atomic desolvation energy. 44 PatchDock is available at http://bioinfo3d.cs.tau.ac.il/PatchDock. The 10 best solutions of PatchDock analysis were selected for further flexible refinement and rescoring analysis through fast interaction refinement in molecular docking (FireDock) algorithm.31,45 This method targets the problem of flexibility and scoring of solutions produced by fast rigid-body docking algorithms. The output provided a list of refined complexes, sorted by a binding energy function, and a 3D visualization for observing and comparing the refined complexes. 45 FireDock is available at http://bioinfo3d.cs.tau.ac.il/FireDock/firedock.html. The docked complexes of native and mutants were analyzed for binding energies, followed by visualization of binding sites using PyMol (http://www.pymol.org).
To identify the binding residues between STK11 and STRAD, we submitted the native and mutants to the protein interface recognition server, solvent accessibility based protein–protein interface identification and recognition (SPPIDER). 46 This server integrates enhanced relative solvent accessibility predictions with high-resolution structural data. This Web server is available at http://sppider.cchmc.org.
Results and Discussion
Retrieval of STK11structure and Mutant Modeling
The native structure (2 WTK) was obtained from PDB database, followed by in silico mutation modeling to generate structures for D194N, E199K, L160P, and Y49D mutants, and subsequently, all the mutant structures were energy minimized.
Identification of Disease-Causing Missense Mutations and Validation
The four mutants, viz., D194N, E199K, L160P, and Y49D, were identified as disease causing through the score of 0.97, 0.91, 0.94, and 0.95, respectively, obtained from NetDiseaseSNP (Table 1). Since the output values were close to 1, all the four selected mutants were considered as disease causing. The mutant D194N changed its amino acid from polar negatively charged to polar neutral, E199K changed from acid to basic, Y49D changed from polar neutral to polar negatively charged, and the mutant L160P retained its basic property.
Scoring of mutants by using NetDiseaseSNP and total energy calculation.

For the validation of this result, we derived the data of corresponding mutation from COSMIC and identified the primary tissue affected by each mutation. The primary tissues affected by the four mutants, viz., D194N, E199K, L160P, and Y49D, were lung, large intestine, cervix, and skin, respectively,11–14 as illustrated in Table 1. The CDS mutation for the above mutants were also obtained from COSMIC, and it was found to be 580G > A, 595G > A, 479T > C, and 145T > G correspondingly.
Ensemble Analysis of Native and Mutant Structures
Divergence of the mutant structure from the native structure could be caused by substitutions, deletions, and insertion, 47 and the deviation between the two structures could alter the functional activity 48 with respect to binding efficiency of the binding partner, which was evaluated by their RMSD, RMSF, and RoG values. The stability of secondary structure elements and conformational changes were assessed by plotting these values obtained from trajectory. To analyze the deviations in conformational space and rigidity of the ensembles, RMSD plot of Ca atoms was computed for native and mutant (D194N, E199K, L160P, and Y49D) structures. The mean RMSD of native was found to be 1.9 Å. On the other hand, all the mutants possessed a higher mean value of RMSD as presented in Table 2 and Figure 1. These results suggested that mutant ensembles comprised structural deviations when compared with native. In addition, RMSF was studied to understand the overall flexibility of the residues of native and mutant. The mean RMSF of native was found to be 0.66 Å, whereas all the mutants possessed increased flexibility in their residues (Table 2 and Fig. 2). Increase in flexibility creates a large loss in enthalpy (weakened native contacts) that is unfavorable for the protein. 49 The analysis of RMSF exposed that the residues 47–97 and 250–276 possessed a comparatively high flexibility change than the other residues in all the mutants. The region 47–97 had involved the 5 binding residues, viz., Leu (67), Thr (71), Leu (72), Cys (73), and Arg (74) 6 for the native–STRAD interaction. Furthermore, RoG analysis disclosed that the three mutants, viz., D194N, E199K, and Y49D, had a less RoG of 18.2918 Å, 18.3645 Å, and 18.3630 Å, respectively, when compared with native 18.4733 Å. Mutant L160P possessed higher value than the native, and it was found to be 18.8869 Å; this could have resulted because of the increase in the flexibility of this mutant (Table 2 and Fig. 3). This, in turn, showed the distinct compactness of mutant structures. Besides, SASA was analyzed, in which, the native exhibited 29,810.50 Å 2 as an average SASA. Three mutants, viz., D194N, E199K, and Y49D, exhibited decreased SASA compared with native, and it was found to be 29,726.60 Å 2 , 29,730.12 Å 2 , and 29,686.71 Å 2 , respectively (Table 2 and Fig. 4), whereas, the mutant L160P showed an increased SASA of 30,070.69 Å 2 . This could have also resulted due to the high RMSF possessed by the mutant L160P, since there is correlation between the flexibility of the residue and SASA. 50 As the flexibility increases, SASA will also increase, which in turn affects the stability of the protein unfavorably.
Trajectory analysis of native and mutants.


RMSD variation of native and mutants.

RMSF of native and mutants. (A) Highly fluctuated region (residues 47–97). (B) Highly fluctuated region (residues 250–276).
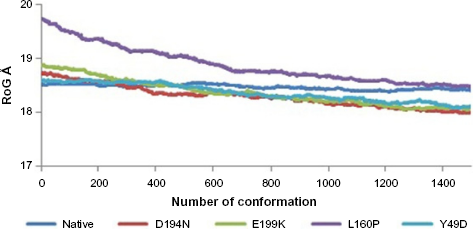
RoG of native and mutants.
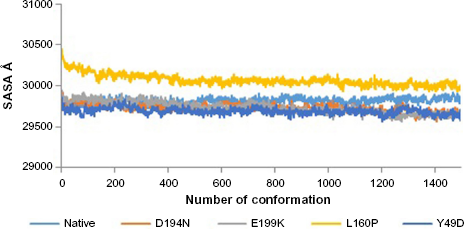
SASA of native and mutants.
Analyzing Structural Events of Global Minima of Native and Mutants
Various types of intramolecular interactions, 51 more specifically hydrogen bonds 52 influence protein folding, 40 which in turn play a major role in the structural property of a protein. The following analyses were carried out between native and mutants using global minimized structures. Hydrophobic, cation-π, and other interactions were contributed in stabilizing proteins. 38 Therefore, intramolecular interactions for native and mutant structures were studied using the PIC program. 36 Different interactions were used to study the variations in structural integrity of the structures, and the results were represented type wise in Table 3. From this result, it was noted that the native structure had a total of 999 interactions, while all the mutants had reduced number of interactions, among which L160P mutant had the lowest with 779 interactions. Reduction in the number of interactions in mutant structures might alter their structural integrity and folding patterns. Therefore, hydrogen bond analysis and dilution patterns were studied in detail to understand the folding patterns of native and mutant structures. Furthermore, energetic contributions of native and mutant structures were analyzed to explore the possible deviations, where the total energy possessed by the native was -16,734.766 kJ/mol, and that of the mutants, viz., D194N, E199K, L160P, and Y49D, were found to be -12,314.974 kJ/mol, -12,326.152 kJ/mol, -10,595.924 kJ/mol, and -13,090.824 kJ/mol, respectively (Table 1). This analysis clearly portrayed that all the four mutants showed increased total energy as compared with native STK11, thus showing alteration in the stability of mutants as compared with native.
Intra molecular interactions of native and mutants.

Identifying the Strength of Hydrogen Bonds, Pattern of Simulated Thermal Denaturation, and Elements of Secondary Structure of Native and Mutants
The pattern of hydrogen bond distribution affects secondary and tertiary structures of proteins. 52 Besides, residues which are far apart in sequence are brought closer by protein folding, as aided by hydrogen bonds. 53 Moreover, changes in folding patterns are known to affect the outline structures of proteins and arrangements of secondary structure elements. 54 Therefore, native and mutant structures were subjected to hydrogen bond analysis and simulated thermal denaturation to analyze the number of hydrogen bonds present in the structure and check how hydrogen bonds were diluted on the application of various energy ranges. The total number of hydrogen bonds present in the native was 284, and most of them (282) reside in the intermediate range, and only 2 were in the long range. Of note, the three mutants, viz., D194N, E199K, and L160P, had less number of total hydrogen bonds of about 273, 283, and 225, respectively, than the native (Table 4). The intermediate range of hydrogen bonds was less in these three mutants, and also, more number of long-range hydrogen bonds was present, when compared with native. The mutant Y49D exhibited more number (292) of hydrogen bonds than the native. Of the 292 hydrogen bonds, about 13 were in the long range and 279 were in the intermediate range. This portrayed that the native possesses strong hydrogen bonds than the mutants, as the length of the hydrogen bonds correlates with the strength of the bond. 37 Native and mutant structures exhibited different patterns of hydrogen bond dilution as noticeable from Table 5 and Figure 5. When the energy of -1 kJ/mol was applied to native and mutants, the remaining hydrogen bonds present in native was 276, whereas the mutants, viz., D194N, E199K, L160P, and Y49D, possessed 265, 274, 226, and 278 hydrogen bonds, respectively. For the energy of-5.8 kJ/mol, the mutant L160P showed complete dilution of hydrogen bonds, whereas the native and the three mutants, viz., D194N, E199K, and Y49D, had remaining hydrogen bonds of 74, 65, 81, and 64, respectively, which, in turn, suggested variations in their folding patterns. In addition, secondary structure elements of native and mutants were visualized and compared with the observed position level variations. It was noted that mutants showed variations in terms of secondary structure elements when compared with native (Fig. 6). These variations could have occurred due to the mutations that might rearrange the packed secondary structure 54 of STK11. The position level variations in secondary structure elements of the mutants were as follows: mutant L160P comprised variations at positions 23–26 (three residues) where, helix was converted to coil. All the mutants showed variations from helix to coil at positions 170–173 (three residues), except Y49D, which showed variation from helix to coil at positions 193–196 (three residues). Three mutants, viz., E199K, L160P, and Y49D, showed variation from coil to helix at positions 203–204, while D194N and E199K showed variations from coil to helix at positions 281–284 (four residues) and L160P showed variation from coil to helix at positions 282–284 (three residues).
Hydrogen bond analysis of native and mutant STK11.

Simulated thermal denaturation of native and mutants.

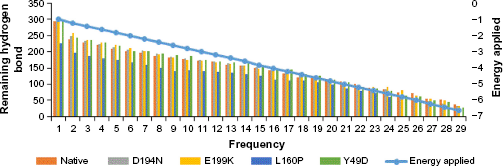
Thermal denaturation of native and mutants with intervals of applied energy.

Secondary structure analysis of native and mutants.
On the whole, the strength of hydrogen bonds, changes in folding patterns, and variations in secondary structure elements gave an idea about the structural changes in the mutant structures. These changes might alter the functional properties of mutant structures that could result in the pathogenesis of the disease, and therefore, functional properties were assessed through docking studies. 55
Computation of Binding Affinity between Native and Mutants
Native and mutant structures were analyzed for their binding affinity toward the partner STRAD. Docking was performed using the PatchDock between STRAD and the global minima of native and mutants of STK11 by specifying the receptor-binding sites such as Arg (74), Cys (73), Leu (72), Thr (71), and Thr (186) and the ligand-binding sites such as Leu (241), His (231), Phe (233), and Gln (251) to determine the binding efficiency in the form of the atomic contact energy (ACE). The 10 best solutions of PatchDock analysis were selected and further analyzed for flexible refinement and rescoring by FireDock. From the analysis, the ACE between STRAD and native STK11 was found to be -3.30 kJ/mol and all the mutants demonstrated low ACE when compared with the native (Table 6). Also, the energy contribution from hydrogen bonding, van der waals force of attraction, and repulsions to the complexes (Fig. 7) of all mutants were varying when compared with native as represented in Table 6. SPPIDER was used to calculate the contacts between the binding residues of native and mutant STK11 with STRAD, and the binding residues were Leu (67), Thr (71), Leu (72), Cys (73), Arg (74), Cys (151), Thr (186), Lys (191), Pro (315), Pro (317), Ile (318), Pro (320), Arg (323), Trp (324), Val (327), Leu (330), and Glu (331). Reduced ACE of mutants toward STRAD might be due to altered RMSF values of binding residues that were present in the highly fluctuated region. Changes in flexibility of these residues might have resulted in improper STRAD binding and eventual loss of functional activity of mutants, which could lead to cancer.
Energy contribution of ACE, attractive and repulsive Vdw and HB in the docked structure of native and mutants with STRAD.

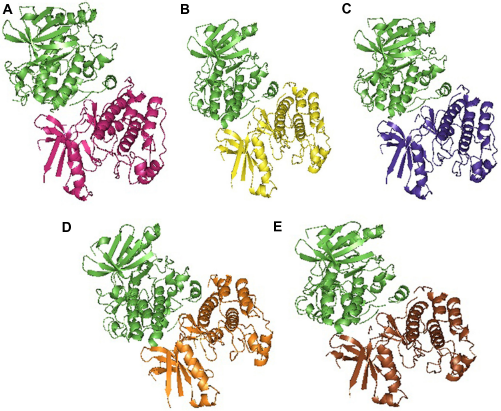
Docked structures of (A) native (magenta) and STRAD (green), (B) D194N (yellow) and STRAD (green), (C) E199K (violet) and STRAD (green), (D) L160P (orange) and STRAD (green), (E) Y49D (brown) and STRAD (green).
Conclusion
Single-nucleotide polymorphism is the most common form of genetic variation, 56 and missense mutations lead to substitution of amino acids, which eventually affected the function of STK11 and were often responsible for causing various types of cancer.11–14 Ensemble analysis of mutants, viz., D194N, E199K, L160P, and Y49D, substantiated considerable variations in terms of RMSD. RMSF of mutants showed variation, especially at the region between 47 and 97, where five binding residues were present, which suggested increased flexibility of residues as compared with native. RoG analysis throughout the ensembles substantiated that the mutants exhibited RoG in the range of 18.2918–18.8869 Å. The SASA analysis of mutants also showed variation from native, possessing SASA in the range of 29,726.60–30,070.69 Å. Furthermore, global minima of native and mutants were used for further analysis. Variations were observed between the native and mutant structures in terms of intramolecular interactions, number of hydrogen bonds, simulated thermal denaturation, and secondary structure elements. Therefore, it was obvious that mutants would have variations in affinity toward partner, which was analyzed through docking studies, where all four mutants had reduced ACE compared with native. This could have eventuated due to the change of flexibility in binding residues and changes in the secondary structure elements. Therefore, it was concluded that mutants, viz., D194N, E199K, L160P, and Y49D, have detrimental effects contributing to the pathogenesis of disease. The study gains interest since the deleterious effects that remained latent within the mutant structures were explored using conformational sampling method that could serve as an alternate to classical molecular dynamics and structural variations of these mutants, which were not reported yet, were explored and might shed more light onto the cancer research community.
Author Contributions
Designed the computational analysis: ML. Analysed and contributed to the writing of the manuscript: DMP. Made critical revision and approved final version: RR. All authors reviewed and approved of the final manuscript.
Supplementary Material
Footnotes
Acknowledgment
The authors are grateful to VIT University for providing necessary facilities and infrastructure to carry out this study.