
Abstract
This paper concerns a study indicating that the expression levels of genes in signaling pathways can be modeled using a causal Bayesian network (BN) that is altered in tumorous tissue. These results open up promising areas of future research that can help identify driver genes and therapeutic targets. So, it is most appropriate for the cancer informatics community.
Our central hypothesis is that the expression levels of genes that code for proteins on a signal transduction network (STP) are causally related and that this causal structure is altered when the STP is involved in cancer. To test this hypothesis, we analyzed 5 STPs associated with breast cancer, 7 STPs associated with other cancers, and 10 randomly chosen pathways, using a breast cancer gene expression level dataset containing 529 cases and 61 controls. We identified all the genes related to each of the 22 pathways and developed separate gene expression datasets for each pathway. We obtained significant results indicating that the causal structure of the expression levels of genes coding for proteins on STPs, which are believed to be implicated in both breast cancer and in all cancers, is more altered in the cases relative to the controls than the causal structure of the randomly chosen pathways.
Keywords
Introduction
There is evidence that similar cancers have many variations at the molecular level, and each has its own clinical course. This is called the
A signal transduction pathway (STP) is a network of information flow in the cells that initiates with a signal outside the cell and results in a cellular response. Many aberrant STPs have been associated with various cancers.4–10 For example, we now know that the ERbB, PI3K–Akt, and Wnt pathways are associated with breast cancer. The signal aberrations associated with a disease often result from one or more mutated genes that code proteins on the pathways. There has been an explosion of new genomic and proteomic datasets providing us with unprecedented and rich resources to reveal the mechanisms of STPs. We have datasets concerning single nucleotide polymorphisms (SNPs), somatic mutations, copy number, methylation levels, and expression levels in both cancerous and non-cancerous tissues.11–13 We have flow cytometry datasets providing us with simultaneous observations of many signaling molecules in a multitude of individual cells.14,15
To develop optimal treatments for cancer patients, it is necessary to address two fundamental issues regarding STPs: (1) the discovery of which STPs are implicated in a cancer or cancer subtype and (2) the prediction of how stimulations and inhibitions will affect the overall activity of the STP.
Using gene expression datasets, a good deal of effort has been devoted to the first issue just mentioned. Initially, techniques such as over-representation analysis16–18 were employed. Such methods ignore the topology of the network, and hence do not account for key biological information. That is, if a pathway is activated through a single receptor and that protein is not produced, the pathway will be severely impacted. However, a protein that appears downstream may have a limited effect on the pathway. Recently, researchers have developed methods that account for the topology of an STP when analyzing gene expression data to determine whether the STP is implicated in a cancer.19–21 Signaling pathway impact analysis (SPIA) 19 is a software package (http://bioinformaticsprb.med.wayne.edu/SPIA) for identifying whether a signaling network is relevant in a given condition that accounts for the topology of the network. However, it is not model based, and does not provide a predictive causal model of an STP. PARADIGM 20 creates a model of a single patient rather than the population, and is able to incorporate copy number variations (CNV) and even mutations. Not being population based, it does not provide an overall causal model of the altered STPs in a given cancer.
To address the second issue (the prediction of how stimulations and inhibitions will affect the overall activity of the STP), we need a causal model of the variables related to an STP. A number of studies22–24 have shown that STPs can be modeled as causal Bayesian networks (BNs) if each node in the network represents the phosphorylation activity of a protein. A strength of BNs is that they represent probabilistic relationships, and therefore they can manage the noise in biological data. A second strength is that they can model the natural causal relationships in biology.
On the one hand, protein phosphorylation assays are slow, relatively expensive, and can be performed for only a tiny but important fraction of the genome. On the other hand, the gene expression level data are widely available because they are inexpensive and genome wide. As noted previously, methods have already been developed that account for the topology of an STP when analyzing gene expression data to determine whether the STP is implicated in a cancer.19–21 However, the correlation of gene expression with activity is not well established. Studies show that the protein expression level (abundance) is often not positively correlated with activity 25 and that the gene expression level is often not correlated with protein abundance. 26 Hence, the gene expression levels might be at most loosely correlated with the activity, which means that the causal structure of an STP might not be represented by the relationships among the gene expression levels. More fundamentally, it is an open question as to whether there are causal relationships among the expression levels of genes coding for proteins on an STP.
We investigated this question. Specifically, the central hypothesis to be investigated in this paper is that the expression levels of genes that code for proteins on a given STP are causally related, and that this causal structure is altered when the STP is involved in a particular cancer. If this hypothesis is correct, using the ample gene expression datasets and BN learning algorithms, we can learn the causal network structure of the gene expression levels in an STP that is altered in a given cancer, and then identify driver genes based on the topology of the network.
The Cancer Genome Atlas (TCGA) makes available a breast cancer dataset that contains data on SNPs and the expression levels of 17,814 genes. There are 529 cases and 61 controls for which this information is available. Using these datasets and BN technology, we investigate the causal structure of genes that code for proteins on 5 STPs believed to be associated with breast cancer, 7 STPs believed to be associated with other cancers, and 10 randomly chosen pathways. We obtain significant results indicating that the causal structure of the STPs, which are believed to be implicated in both breast cancer and all cancer, is more altered in the cases relative to the controls than the causal structure of the randomly chosen pathways.
Method
As our method applies BNs to modeling STPs, we first review both of these.
BNs.
BNs27–29 are increasingly being used for uncertain reasoning and machine learning. A BN consists of a directed acyclic graph (DAG)
Figure 1 shows a BN representing the causal relationships among variables related to lung disorders. In this BN,

A BN representing a subset of the variables related to lung disorders. There is an edge from node
A BN DAG model consists of a DAG
In the constraint-based approach,
30
we learn a DAG model from the conditional independencies that the data suggest are present in the generative probability distribution 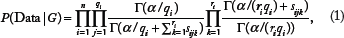
Many biological processes have been modeled using BNs including molecular phylogenetics, 33 gene regulatory networks,34–36 genetic linkage, 37 genetic epistasis,38–42 and STPs.22–24 Figure 2 shows a BN representing a small gene regulatory network.

A BN for a small gene regulatory network (based on a figure in Ref 33). Only the conditional probability distribution for node S is shown. Each variable is continuously distributed, and defined to be “high” if its value is higher than 1 and “low” if its value is less than 1. The notation ρ(
STPs Modeled as BNs.
An STP is a network of intercellular information flow initiated when extracellular signaling molecules bind to cell-surface receptors. The signaling molecules become modified, causing a change in their functional capability and affecting a change in the subsequent molecules in the network. This cascading process culminates in a cellular response. Consensus STPs have been developed based on the composite of studies concerning individual STP components. Figure 3 shows part of the consensus STP of human primary naive CD4 T cells, downstream from CD3, CD28, and LFA-1 activation. Kyoto Encyclopedia of Genes and Genomes (KEGG) 43 has a collection of manually drawn pathways representing our knowledge of about 136 pathways. STPs are not thought to be stand-alone networks, but rather they have inter-pathway communication. 44

A portion of the consensus STP of human primary naive CD4 T cells, downstream from CD3, CD28, and LFA-1 activation. Arcs are used to illustrate connections between signaling molecules. In some cases, the connections may be indirect and may involve specific phosphorylation sites of the signaling molecules. MAPKKK appears twice because MEK4/7 and MEK3/6 each have a MAPKKK that is its activator. This figure is based on a figure in Ref. 14; see that paper for more details.
If we represent the phosphorylation level of each protein in an STP by a random variable and draw an arc from
As discussed in the Introduction, gene expression level seems to be at most loosely correlated with activity. So, if there are causal relationships among the expression levels of genes coding for proteins on an STP, the BN representing these relationships may not represent the biological flow of an STP. This means it would be difficult to learn STPs from the gene expression levels. However, if our goal is to investigate how variables concerning known STPs are modified in tumors, not to learn the structure of unknown STPs, then the causal structure of the gene expression levels in tumors can provide us with important information. As also mentioned in the Introduction, it is an open question as to whether there are even causal relationships among the expression levels of genes coding for proteins on an STP. This paper investigates this question.
Identifying Aberrant STPs Using BNs and Gene Expression Level Data.
In what follows, for simplicity we will say that a gene coding for a protein on an STP is on the STP itself. We assume that we have two sets of data. The first set contains the gene expression levels of all (at least most) genes in a set of cases (tumors) and the second set contains the gene expression levels of all genes in a set of controls. Let STPX be an STP we are investigating, Data1 be the data concerning the cases for genes on STPX, and Data2 be the data concerning controls for genes on STPX.
There are two models. Model 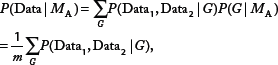
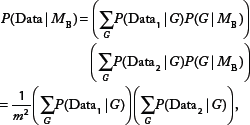

An alternative method would be to approximately learn the most likely DAG model 
The larger the value of
Evaluation Methodology
It is difficult to assess a pathway analysis model or methodology using real data because the ground truth is not known. In the absence of a gold standard, we can perform our analysis based on the existing biological knowledge. Hence, to investigate whether the causal structure of the expression levels of genes on an STP is altered when the STP is involved in cancer, we compared results obtained using the breast cancer data for 5 STPs implicated in breast cancer, 7 STPs implicated in other cancers, and 10 random pathways. We investigated STPs implicated in other cancers because it is believed that there are commonalities across tumor lineages.
45
The pathways investigated are listed in Table 1. The first column lists the five STPs believed to be implicated in breast cancer. The PI3K pathway is one of the most important pathways in cancer metabolism in general, and has recognized as an important target in breast cancer management for years.
46
Hyperactive Wnt signaling has been shown to contribute to cancer in a wide range of human tissue, and Wnt genes have been identified as oncogenes in mouse mammary tumorigenesis.
47
Over-expression of the
Pathways investigated.

The cancer genome atlas (TCGA) makes available a breast cancer dataset that contains data on SNPs and the expression levels of 17,814 genes. There are 529 cases and 61 controls for which this information is available. Using the KEGG database, we identified all the genes related to each of the 22 pathways. We extracted gene expression profiles for the 529 breast cancer patients and 61 controls in the TCGA database. By mapping the gene names of the genes in the gene sets identified using the KEGG pathways and the gene names in the TCGA data, we were able to extract the gene expression profiles for each of the 22 pathways for the 529 patients and 61 controls. All the expression levels were discretized to values
All experiments were run using a Dell PowerEdge R515, which has two AMD Opteron™ 4276HE, 2.6 GHz, 8C, Turbo CORE, 8M L2/8M L3, 1600 MHz Max Mem single processors.
Results
Table 2 lists the pathways, along with their Bayes factors, in a decreasing order. It is notable that PI3K, which is “probably one of the most important pathways in cancer metabolism and growth,” 52 scored much higher than all other pathways. The Wnt and ErbB pathways are also near the top of the list. However, the Notch and Hedgehog pathways are not. In general, however, the cancer-related pathways are concentrated at the top of the list. Figure 4 shows the average Bayes factor and standard error for each of the three categories.
Bayes factors for 22 pathways. There is an “X” if the pathway is implicated in breast cancer or any cancer.


Average Bayes factors and standard error for breast cancer pathways, all cancer pathways, and other pathways, when causation is modeled.
Table 3 shows the

The possibility exists that these significant results were obtained simply because the genes are over or under expressed in cancer-related STPs and the causal structure is not relevant. To test this possibility, we redid the study with all the BNs constrained to having no causal edges. Table 3 shows the resultant
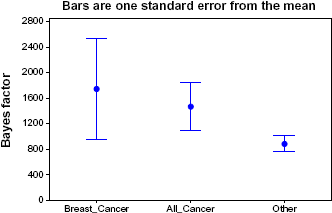
Average Bayes factors and standard error for breast cancer pathways, all cancer pathways, and other pathways, when causation is not modeled.
All networks learned are fairly complex. As an example, Figure 6 shows the network learned from cases for the ErbB pathway.

The causal BN learned from breast cancer cases for the ErbB pathway.
Discussion
We analyzed 5 STPs associated with breast cancer, 7 STPs associated with other cancers, and 10 randomly chosen pathways. Based on modeling the relationships among the expression levels of genes on the pathways as causal BNs, we obtained results indicating that the causal structure of the cancer-related STPs is significantly more altered in breast cancer tissue than the randomly chosen pathways. These results support that the expression levels of genes on STPs are causally related and that this causal structure is altered in the tumorous tissue when an STP is involved in cancer.
These results are significant for a number of reasons. First, we can use the methodology to develop a method for investigating whether an STP is involved in cancer, which can be compared to the existing methods.30,31,53 Second, these results open up a promising area of future research involving the use of BN technology to model the causal relationships among the expression levels of genes on an STP. Using such a network, we can learn possible driver genes, and the effect of genetic variants on these driver genes and therefore on the network. Such investigations would enable us to better identify therapeutic targets in a patient-specific fashion.
In future research, we can implement the Bayes factor calculation (Equation 1), and see if it yields better results than the approximation used in the given studies. Furthermore, we can develop and implement a method that better learns the causal edges among the genes in the STP. Rather than just learning a single highly likely model using a package like HUGIN, we can do approximate model averaging to learn the strength of the edges. Finally, we can develop and test an entire BN that contains both expression levels and genetic causes of expression levels.
Conclusion
We conclude that our study supports that the relationships among the expression levels of genes on an STP can be modeled using a causal BN, and that this network is altered in the tumorous tissue. This result opens up new avenues for identifying driver genes on STPs.
Author Contributions
XJ conceived and designed the experiments. DX processed the data, developed the datasets representing the pathways, and analyzed the data. RN wrote the first draft of the manuscript. XJ contributed to the writing of the manuscript. RN and XJ jointly developed the structure and arguments for the paper. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
References
 ghici S. et.al.
ghici S. et.al.