
Abstract
The Ras association domain family 1 (RASSF1) gene is a Ras effector that plays an important role in carcinogenesis. We have previously shown that silencing of RASSF1C decreases and over-expression of RASSF1C increases cell proliferation, migration, and attenuates apoptosis of breast cancer cells in vitro. To further confirm our working hypothesis that RASSF1C may play a role as a growth promoter, we have tested the growth of human breast cancer cells stably over-expressing RASSF1A or RASSF1C in nude mice. Our studies show that breast cancer cells over-expressing HA-RASSF1A developed significantly smaller tumors and cells over-expressing HA-RASSF1C developed significantly larger tumors compared to control cells expressing the vector back bone. We have confirmed the expression of HA-RASSF1A and HA-RASSF1C in tumor tissue using RT-PCR, western blotting and immunohistochemical analyses using HA-antibody. Together, our previous in vitro and current in vivo findings further support our hypothesis that RASSF1C, unlike RASSF1A, is not a tumor suppressor and rather it appears to function as tumor growth promoter in breast cancer cells.
Introduction
The RASSF1 gene encodes seven isoforms (A-G) derived by different promoter usage and alternative mRNA splicing.1,2 RASSF1A and RASSF1C are the two major isoforms encoded by the RASSF1 gene. RASSFIA is a protein of 340 amino acids and RASSF1C is a protein of 270 amino acids with identical C-termini, but with different N-termini.1,2
RASSF1A is well studied and unequivocally functions as a tumor suppressor. RASSF1A mRNA expression is epigenetically inactivated by cytidine methylation in many types of human solid tumors including breast, lung, liver, and prostate,1–10 while RASSF1C is typically fully expressed in tumors. Furthermore, restoring RASSF1A expression decreases colony formation, reduces tumor growth in nude mice, and induces cell cycle arrest by blocking cyclin D accumulation.2,8,11,12 In addition, RASSF1A –/– and RASSF1A +/– mice exhibit enhanced tumor multiplicity and tumor size compared to wild type animals upon exposure to the chemical carcinogens benzo(a) pyrene and urethane. 13 Re-expression of RASSF1A in cancer cell lines decreased cell proliferation, arrested cell cycle, and induced cell apoptosis and senescence in part through the modulation of MST2, MEK/ERK, and PI3K/Akt activities.14–16
In contrast to RASSF1A, RASSF1C is expressed in almost all human solid tumors including breast cancer. The majority of published literature indicates that RASSF1C has no tumor-suppressor activity.2,9,11,12,17 However, some reports suggest that RASSF1C may function as a tumor suppressor in ovarian, prostate, renal cancer cells.18–20 We have previously shown that silencing of RASSF1C decreases, and over-expression of RASSF1C increases, cell proliferation in breast, lung, and osteosarcoma cancer cells in vitro suggesting a growth promoting role for RASSF1C.21–23 We have also shown that RASSF1C increases cell migration, and attenuates apoptosis of breast cancer cells in vitro. 23 Therefore, the purpose of this study is to further confirm our in vitro data that RASSF1C, unlike RASSF1A, acts as tumor growth promoter in breast cancer cells in vivo. In this paper we report on our in vivo studies involving human breast cancer cells stably over-expressing RASSF1A or RASSF1C in nude mice.
Materials and Methods
Cell culture
The human breast cancer cell line T47D stably over-expressing HA-RASSF1A or HA-RASSF1C was developed as previously described. 20 The vector is a Murine Leukemia Virus (MLV) based retroviral vector that was developed in house as previously described. 23 T47D cells over-expressing HA-RASSF1A (T47D-1A), HA-RASSF1C (T47D-1C), or the MLV back bone (T47D-BB) were grown in RPMI-1640 medium supplemented with 10% calf bovine serum and 0.2 units/mL insulin.
Experimental animal model
Five-week old athymic female nude (Crl: CD-1Fox-n1nu, Charles River) mice were used, and 3x106 cells at 70% confluence (cells in 200 ul of RPMI serum free medium) were injected subcutaneously into the flank, one injection per animal. Four mice were injected with T47D-BB, four mice were injected with T47D-1C, and four mice were injected with T47D-1A cells. Mice were monitored and tumors were measured twice weekly using a caliper for 6–8 weeks. After 8 weeks, tumors were harvested and tumor tissue was fixed in 4% paraformaldehyde and used for mRNA isolation and protein lysate preparation. Tumor volume for each group (n = 4) was calculated using the formula: 1/2(length × width 2 )21,22 and all animals injected grew tumors that varied in size.
Frozen tumor tissue was grounded to a powder using a mortar and pestle and ground tissue was used for isolating mRNA using absolutely RNA Microprep Kit (Stratagene, La Jolla, CA) and for making cell lysates using RIPA buffer supplement with protease inhibitors. The animal study protocols were approved by the Loma Linda VA Medical Center IACUC.
qRT-PCR analysis
Expression of RASSF1A and RASSF1C in tumor tissue derived from cell lines was assessed by qRT-PCR using RASSF1A and RASSF1C specific primers. The PCR reactions were run using the following protocol: (1) incubate at 95 °C for 10 min, (2) incubate at 95 °C for 15 sec, (3) incubate for 30 sec, (4) go to line 2 for 39 more cycles, (5) melting curve from 60 °C to 95 °C, read every 1.0 °C, (6) incubate at 10 °C forever. qRT-PCR reactions were carried out in triplicate and the fold change was calculated using the 2–δδCT method. 26 RASSF1A forward primer is 5′GGCGTCGTGCGCAAAGGCC and RASFF1C-forward primer: is 5′CTGCAGCCAAGAGGACTCGG 3′, and the reverse primer for both RASSF1A and 1C is 5′GGGTGGCTTCTTGCTGGAGGG3′. Cyclophillin (housekeeping gene) forward primer 5′GCATACAGGTCCTGGCATCT3′ and Cyclophillin reverse primer 5′TCTTGCTGGTCTTGCCATTC3′.
Western blot analysis
HA-RASSF1A and HA-RASSF1C expression in tumor tissue was assessed by Western blot analysis using the following anti-bodies, mouse Mono anti-HA (Mon HA.11(16B12) Covance, Berkeley, CA), Mouse monoclonal p-ERK (E-4) (sc-7383, Santa Cruz, Biotechnology, CA), Mouse anti-panERK (Cat. 610123, BD Transduction Laboratory), and Goat anti-Mouse and Donkey anti-Rabbit fluorescently labeled secondary antibodies (IRDye@680926-32220, LI-COR Biosciences) were used. Blots were incubated with appropriate primary antibody at a 1:1000 dilution in LI-COR blocking buffer (LI-COR Biosciences) overnight at 4 °C on a rotary shaker. After primary antibody incubation, blots were washed three times with 1XPBS with 0.1% Tween 20 (1XTPBS) for five minutes before adding the secondary antibody at 1:1000 dilution for one hour incubation at room temperature. After secondary antibody incubation, blots were washed with 1XTPBS) three time for five minutes and blot were scanned using Odyesey scanner. Western blots were repeated three times.
Histology and immunohistochemistry
Immunohistochemical staining of T47D xenograft sections for HA-RASSF1A and HA-RASSF1C fusion proteins was carried out with the diaminobenzidene (DAB) method. Five μm sections cut from paraffin-embedded blocks were deparaffinized, and 3% H2O2 was applied for 30 min at room temperature. Sections were then washed three times with H2O and blocked with 20% normal horse serum (Vector Laboratories) for 30 min at 37 °C. Horse serum was removed and 1:1000 mouse anti-HA antibody (Covence) was applied for 1 h at room temperature. Sections were washed three times for 5 min each with PBS/Tween, and 1:1000 biotinylated anti-mouse IgG was applied for 30 min and sections were washed three times with PBS/Tween. Horeseradish peroxidase strepavidin (Vector Laboratories) was then applied for 20 min at room temperature at 1:200 dilutions. Sections were washed three times with PBS/Tween and DAB chromogen (Biocare Betazoid DAB Chromogen) for 5 min at room temperature and sections were washed three times with H2O and counterstained with Hematoxylin (Harris Hematoxylin, Fisher Scientific).
Statistical analysis
Tumor volumes of xenografts from animals injected with T47D-HA-RASSF1A (T47D-1A), T47D-vector backbone (T47D-BB), and T47D-HA-RASSF1C (T47D-1C) human breast cancer cells were analyzed using the t-test.
Results
RASSF1C enhances breast cancer tumor growth
T47D cells over-expressing HA-RASSF1A (T47D-1A), HA-RASSF1C (T47D-1C) or the MLV back bone (T47D-BB) were injected subcutaneously in athymic nude mice to form tumors. Tumors were measured for 59 days after injection (Fig. 1A). Figure 1A shows that breast cancer cells over-expressing RASSF1A (T47D-1A) and control cells (T47D-BB) developed significantly smaller tumors compared to cells over-expressing RASSF1C (T47D-1C). Figure 1B shows representative mice growing T47D-1A, T47D-BB, and T47D-1C xenografts. Our findings are consistent with our hypothesis and clearly suggest that RASSF1C appears to function as a growth promoter in breast cancer cells.
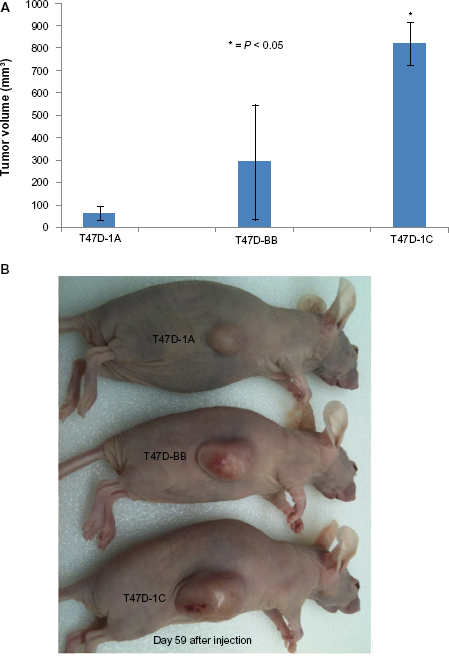
(
RASSF1A and RASSF1C are stably expressed in xenograft tissue
In order to determine if HA-RASSF1A and HA-RASSF1C were stably over-expressed in the appropriate xenograft tissue over the course of the experiment, we performed qRT-PCR which showed that RASSF1A and RASSF1C were expressed ≫ 15 fold higher compared to control. We also performed Western blot (Fig. 2) and immunohistochmistry(Fig. 3). Each of these methods clearly showed that both HA-RASSF1A and HA-RASSF1C were stably over-expressed in the appropriate tumors.
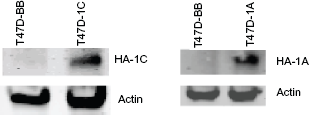
Western blot analysis of protein lyastes from xenografts derived from tumors of T47D-BB, T47D-1A, and T47D-1C.
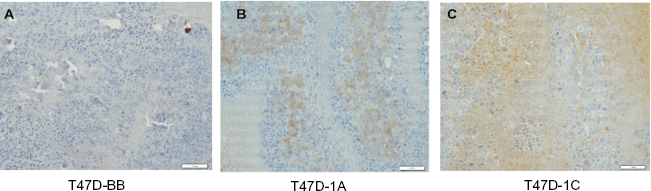
Immunohistochemical analysis was carried out for HA-RASSF1A and HA-RASSF1C fusion proteins in T47D-derived tumors sections. The diaminobenzidene (DAB) method was used, and the HA-tag antibody was used as the primary antibody. (
RASSF1C over-expression increases ERK1/2 phosphorylation in vivo
Because previous in vitro work suggested that RASSF1C may function via activation of the MEK/ERK pathway, 18 we measured activated ERK1/2 levels in tumors over-expressing T47D-BB and T47D-1C. Total cell lysates from subcutaneous tumors were subjected to Western blot analysis using antibodies specific for phosphorylated ERK1 and ERK2 (Fig. 4). The level of phosphorylated ERK1/2 in T47D-1C tumor tissue is much higher compared to that in T47D-BB tumor tissue. It should be noted that RASSF1A over-expression did not affect ERK1/2 phosphoryaltion(data not shown).
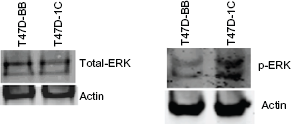
ERK1/2 activation was assessed in T47D-BB and T47D-1C derived tumor tissues.
Discussion
In previous studies we have shown that RASSF1C over-expression increases, and silencing of RASSF1C expression decreases, cell proliferation of breast and lung cancer cells in vitro.22,23 We have also demonstrated that over-expression of RASSF1C enhances breast cancer cell migration and attenuates apoptosis, suggesting a growth promoting function(s) for RASSF1C. 23 Our previous in vitro findings suggest that RASSF1C and RASSF1A appear to have antagonistic functions in breast, lung, and bone cancer cells.22,23 To further confirm the impact of RASSF1C on cancer cell growth, we have carried out animal studies using breast cancer cells stably over-expressing RASSF1C.
Our in vivo studies show that breast cancer cells stably over-expressing RASSF1C developed significantly larger tumors compared to cells over-expressing the vector back bone or over-expressing RASSF1A (Fig. 1). Our in vivo findings are consistent with our previously published in vitro work suggesting a growth promoting function for RASSF1C, and our findings are the first to demonstrate that over-expression of RASSF1A and RASSF1C have opposite effects on breast tumor growth in vivo. We confirmed the expression of HA-RASSF1A and HA-RASSF1C at protein levels (Figs. 3 and 4) in T47D-1A and -1C derived tumor tissues to demonstrate that both proteins were stably expressed. Our previous in vitro work and current in vivo work underscores a growth promoting function for RASSF1C which is opposite to that reported for ovarian, prostate, renal cancer cells.21–23 As to why RASSF1C may exert stimulatory function in some human cancers such as osteosarcoma, breast cancer, and lung cancer cells,21–23 and may exert inhibitory functions in prostate, kidney, and ovarian cancer cells18–20 is still to be determined. One potential explanation for such opposite effects of RASSF1C in different types of human cancers may reflect tissue-specific actions of RASSF1C.
It will now be important to elucidate the mechanism(s)/pathway(s) through which RASSF1C exerts its growth promoting effects on breast cancer cells. Previously we have found that cancer cells over-expressing RASSF1C exhibited increased phosphorylation of ERK1/2. 22 In the current study, we measured the levels of phosphorylated ERK1/2 in tumors over-expressing RASSF1C. We found that tumors derived from cells over-expressing RASSF1C showed increased levels of phosphorylated ERK1/2 compared to control (Fig. 4), suggesting that RASSF1C may exert its functions in part through the activation of the MEK/ERK1/2 pathway in vivo.
Obviously, more work is required to prove if there is indeed a link between RASSF1C activities and the activation of the MEK/ERK pathway, and we have studies in progress that are designed to address this question. In conclusion, although our previous and current findings provide evidence that RASSF1C is not a tumor suppressor, more insightful mechanistic work is required to demonstrate that RASSF1C, unlike RASSF1A, has growth promoting activities in breast cancer cells.
Author Contributions
MR participated in the design of the study, contributed to data analysis, and drafting of the manuscript. RA performed animal injections and tumor measurements and analysis. MA carried tissue culture work, RNA work, RT-PCR, western blot analysis. SB carried tissue culture and gene cloning work. SC prepared and viral vectors and viral cell transduction. NL performed histology and immunohistochemistry work. SM participated in drafting the manuscript. YA designed and supervised the study, carried out the gene silencing and over-expression work, and contributed to data analysis and drafting of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by a grant from the Department of Surgery, Loma Linda University School of Medicine.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgment
The work was carried out at the Loma Linda VA Medical Center.