
Abstract
Pancreatic adenocarcinoma is associated with advanced presentation and poor survival. Currently approved therapies have minimal effect on patient survival. Pancreatic adenocarcinomas have a high incidence of activated K-RAS, which may confer resistance to epidermal growth factor receptor (EGFR) inhibitors. Mixed lineage kinase-3 (MLK3) is a MAP3K that activates multiple MAPK pathways. The role of MLK3 in the pathophysiology and resistance to therapy of pancreatic adenocarcinoma has not been investigated. MLK3 is over expressed in pancreatic cancer cell lines compared to an immortalized pancreatic epithelial cell line. The requirement of MLK3 for cell proliferation and survival of pancreatic cancer cell lines, PANC-1 and MiaPaCa-2, was investigated using RNA interference (siRNA) and MLK inhibitor, K252a, alone or in conjunction with the EGFR inhibitor, Compound 56. Ablation of expression of MLK3 via siRNA-mediated gene silencing and pharmacological inhibition of MLK3 by K252a each decreased cell viability in both pancreatic cancer cell lines, with a concurrent decrease in the activation of ERK, JNK and AKT. Concomitant inhibition of EGFR and MLK3 induced apoptosis, as evidenced by increased cleavage of PARP and caspase-3. These results suggest that MLK3 plays an important role in survival and proliferation of pancreatic cancer cell lines and that inhibition of MLK3 may enhance the therapeutic efficacy of EGFR inhibitors in the treatment of pancreatic cancer.
Introduction
Pancreatic adenocarcinoma constitutes about 90% of pancreatic cancers in humans. Pancreatic cancer is the fourth leading cause of cancer deaths in the United States. 1 Around the globe, over 200,000 individuals will be afflicted by this disease annually. Each year in the United States, nearly 38,000 individuals are diagnosed with pancreatic cancer and over 34,000 succumb to this condition, with a median survival of about 6 months. The 5-year overall survival rate for pancreatic cancer patients is only 5%, due to the advanced stage at presentation and the lack of effective treatments. 2 Novel diagnostic and therapeutic strategies are desperately needed to improve the survival of these patients. Recent data suggest that the molecular portrait of pancreatic cancer is rather complex, involving multiple genetic abnormalities and epigenetic changes. 3 Targeted therapy directed against critical, aberrant signaling pathways represents a promising area of cancer research. However, such therapies have not yet been efficacious, perhaps due to activation of other compensatory signaling pathways that promote proliferation and/or survival. It is important to identify additional signaling pathways that may be exploited to increase the effectiveness of targeted agents for the treatment of pancreatic cancer.
Despite the promise of targeted therapeutic approaches in pancreatic cancer, currently approved therapies, including epidermal growth factor receptor (EGFR) inhibitors, have minimal effect on patient survival. 4 EGFR overexpression is detected in up to 90% of pancreatic tumors, including pancreatic adenocarcinoma. 5 EGFR regulates intracellular signaling processes via the RAS/Raf/MAP kinase pathway, the phosphatidyl-inositol-3-phosphate kinase (PIP Kinase/PI-3K)/AKT pathway, and the JAK/STAT pathway. 6 Greater than 90% of human carcinomas of the exocrine pancreas have oncogenic, activated K-RAS. 7 Constitutive signaling inititiated by mutated K-RAS may bypass inhibition of EGFR, resulting in resistance to EGFR inhibitors, as has been shown in colorectal cancer.8,9
An interesting family of proteins, which might regulate effector pathways of K-RAS, are the mixed-lineage kinases (MLKs).
10
MLKs are serine/threonine protein kinases that function as MAPK kinase kinases (MAP3K), to activate c-jun amino-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p-38 pathways.10–13 As shown in Figure 1, MLK3 exerts its function presumably by directly phosphorylating MKK4/7 (for the JNK pathway) and MKK3/6 (for the p38 pathway).12,14 MLK3 is proposed to act as a scaffold in B-Raf mediated ERK activation.
15
Silencing of
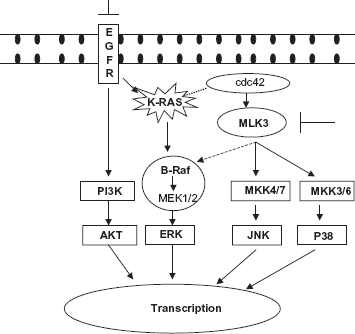
Schematic representation of working hypothesis that simultaneous inhibition of EGF receptor (EGFR) and mixed lineage kinase-3 (MLK3) induces decrease in cell viability in pancreatic cancer cells.
Although MLK3 was previously implicated in neuronal apoptosis, 16 there are several lines of evidence to suggest that MLKs, including MLK3, might contribute to cancer progression. Overexpression of wild type MLK3 can transform NIH3T3 cells. 17 In addition, a relatively high level of MLK3 has been noted in breast cancer cell lines as compared with nontumorigenic mammary epithelial cell lines (Gallo lab, in review). Overexpression of an MLK-like protein, MLTK, was shown to be sufficient to induce cell transformation and formation of fibrosarcoma in a nude mouse model. 18 Pharmacological inhibition of MLK also was shown to cause cell cycle arrest in HeLa cells, preventing cell proliferation. 19 These studies suggest that MLK family proteins, including MLK3, might have a role in the cellular transformation leading to a cancer phenotype.
Since MLK3 regulates cellular functions via known effectors of the K-RAS pathway, including JNK, ERK, and p38, we hypothesized that MLK3 might be a potential cancer therapy target in combination with EGFR inhibitors in pancreatic cancer (Fig. 1). Herein, we report that MLK3 expression is significantly higher in human pancreatic cancer cell lines compared to a normal pancreatic ductal epithelial cell line. Inhibition of MLK3 expression using RNA interference or MLK3 activity by pharmacological inhibition, led to a decrease in pancreatic cancer cell viability. Combined inhibition of EGFR and MLK3 decreased cancer cell viability, compared to either agent alone. Based on these results, we propose that MLK3 may be a potential target for therapeutics in pancreatic cancer that could be used in combination with conventional chemotherapeutic agents and/or with other targeted therapies including EGFR inhibitors.
Materials and Methods
Cell lines
Two well-established pancreatic cancer cell lines, PANC-1 and MiaPaCa-2 (both with
Pharmacological agents
K252a ((9S, 10R, 12R)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-meth yl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6] benzodiazocine-10-carboxylic acid methyl ester, C27H21N3O5) (EMD, Darmstadt, Germany) was used to pharmacologically inhibit MLK3. K252a was dissolved in dimethyl sulfoxide (DMSO). A small molecule EGFR inhibitor, compound 56 (CMP56, 4-[(3-Bromophenyl)amino]-6,7-diethoxyquinazoline), was purchased from EMD (Darmstadt, Germany). CMP 56 was dissolved in DMSO at the appropriate concentrations.
Antibodies
EGFR, ERK, phospho-ERK, AKT, phospho-AKT(473), Poly (ADP-ribose) polymerase (PARP), caspase 3, JNK, phospho-JNK antibodies were obtained from Cell Signaling Technology (Boston, MA). Antibody to phospho-EGFR (T1173) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). MLK3 antibody was produced previously. 20
RNA interference (siRNA)
Synthetic double-stranded RNA (dsRNA) oligonucleotides targeting
Cell treatment
Pharmacological agents were added to culture medium at the indicated final concentrations, combinations and time course. Cells were plated in duplicate in 6-well plates, and transfected as described above. In transfection experiments, medium was supplemented with inhibitor (K252a) 24 h after transfection, for an additional 24 h. Negative controls lacking inhibitor were set up in all experiments. All the experiments were repeated at least twice.
Western blotting
Cells were washed with ice-cold phosphate-buffered saline, and whole cell extracts were prepared in protein lysis buffer (100 mM Tris HCl (pH 7.4), 20% SDS, 10 mM EDTA, 5 mM EGTA) supplemented with protease inhibitor cocktail 1:200 dilution (Sigma-Aldrich, St. Louis, MO) and 2.0 mM sodium orthovanadate. Proteins were resolved by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in 5% milk, the blots were incubated with antibodies using the following dilutions MLK3 (1:1000), p-EGFR, p-AKT (Santa Cruz), pERK, p-JNK, PARP, caspase 3 (1:250) (Cell Signaling) and actin 1:2000, (Sigma-Aldrich, St. Louis, MO). After washing with Tris-buffered saline containing the detergent 0.01% Tween 20, the membranes were incubated with anti-mouse or anti-rabbit IgG-horseradish peroxidase conjugate antibody (Biorad) at a 1:5000 for 1 h at room temperature and developed using an ECL detection kit (GE Healthcare, Piscataway, NJ). Semi-quantitative measurements of western blots were performed using densitometric analysis (ImageJ gel analysis program, http://rsbweb.nih.gov/ij/). The density of the band corresponding to the protein of interest was normalized to that of the actin band.
MTT assays
Cell viability was determined using the MTT assay as per the manufacturer's (Sigma-Aldrich) instructions. Briefly, cells were seeded in 96-well plates at a density of 1 × 105 cells/plate, grown overnight and exposed to CMP56 or K252a for 24–72 h. After the incubation, 10 μl thiazolyl blue tetrazolium bromide (MTT), was added to the wells. After 3 h in a CO2-incubator, formazan products were solubilized with 100 μl detergent for a total volume of 300 μl and the optical density was measured at 570 nm. To determine the concentration at which CMP56 and/or K252a substantially decreased cell viability, graded concentrations were added to duplicate plates using the formula (C-T/C) × 100, where T is the optical density of the test plate sample and C is control optical density. All points were read as mean ± SD of readings of three samples.
Caspase 3 activity
The PathScan Sandwich Elisa Kit (Cell Signaling Technology) was used to detect levels of cleaved caspase-3. Total caspase-3 antibody was coated onto a micro-well plate and, following incubation with cellular lysates, the total caspase-3 protein was captured. Following extensive washing, a biotinylated cleaved caspase-3 antibody was added to detect the captured cleaved caspase-3 protein. An HRP-linked streptavidin was then used to recognize the bound detection antibody and an HRP substrate was added to develop color. The magnitude of optical density for the developed color was proportional to quantity of cleaved caspase-3 protein.
Statistical analysis
Quantitative results were expressed as mean ± SEM. Statistical analysis was performed using a two-tailed unpaired t test (between two groups) or a one way analysis of variance (ANOVA) with the computer software SPSS. P < 0.05 was considered statistically significant.
Results
Expression of MLK3 in normal and malignant pancreatic cell lines
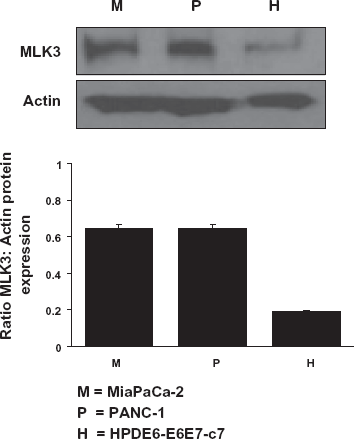
The expression of MLK3 in cellular lysates derived from both “normal” and maligant pancreatic cell lines was semi-quantitatively measured after western blot analysis using densitometry (Optical Density units). As shown in Figure 2, the MLK3 protein level is significantly (nearly 3-fold) higher in both PANC-1 (P) and MiaPaCa-2 (M) pancreatic cancer cell lines than in the immortalized pancreatic epithelial cell line (H).
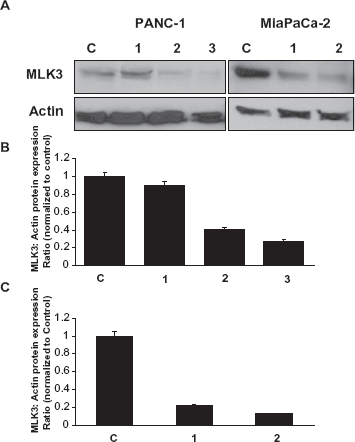
Inhibition of MLK3 expression in pancreatic cancer cell lines
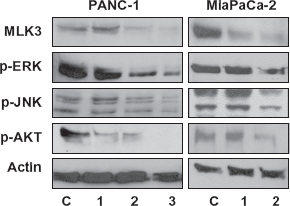
Using RNA interference, we successfully reduced MLK3 expression in both PANC-1 and MiaPaCa-2 cells. As expected, expression of MLK3 protein in
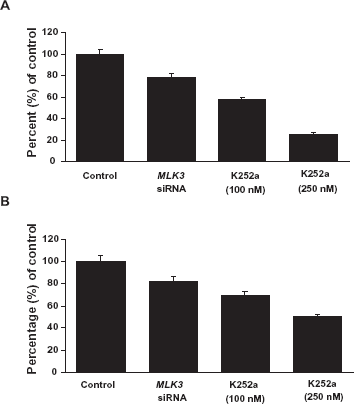
Inhibition of MLK3 expression and activity leads to decreased cell proliferation
In order to understand the role of MLK3 in cell survival and proliferation in pancreatic cancer cell lines, we investigated the fate of cancer cells after inhibiting MLK3, either by decreasing its expression via siRNA or inhibiting its activity using K252a, which effectively inhibits MLK3 activity.
21
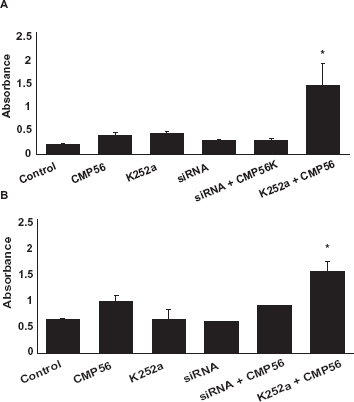
As shown in Figure 4, cell proliferation was reduced significantly by both
Inhibition of MLK3 causes pancreatic cancer cell apoptosis possibly through blockade of JNK and ERK pathways
To better understand the mechanisms underlying decreased cell proliferation due to MLK3 inhibition, we hypothesized the involvement of MAPKs as well as the mitochondrial apoptotic pathway in this process. To test this, the activation status of various MAPKs was assessed by western blotting with appropriate phospho-specific antibodies, after treating the pancreatic cancer cell lines with
Discussion
Pancreatic cancer is one of the most aggressive forms of cancer. Standard gemcitabine-based regimens result in an overall survival of less than a year.
22
With advances in understanding the biology of this aggressive cancer, several targeted therapies are under evaluation,
23
but these efforts are far from clinical use. One important target of recent interest in pancreatic cancer is the EGFR pathway. Preclinical models interrupting EGFR signaling demonstrated a significant antitumor effect.
24
However, results from clinical trials, thus far, show only a modest benefit for such therapies in patients with pancreatic cancer when anti-EGFR agents were combined with gemcitabine.
4
This could be secondary to constitutive activation of signaling molecules downstream of EGFR, and/or to redundant and cross talk pathways. It is well known that activating
Our approach for a combined targeted strategy against proliferative and apoptotic signaling was based on the known molecular effects of EGFR inhibitors in pancreatic cancer 24 and known effects of MLK3 inhibition in cancer cell lines.15,18 In a seminal study by Chadee et al, 15 it was noted that MLK3 is important for proliferation in colorectal cancer cells harboring activating K-RAS mutation, but not those with B-Raf mutations. By using RNA interference of MLK3, they demonstrated marked suppression of cell proliferation in several cancer cell lines including the human adenocarcinoma cell line HCT15, which harbors an activating G13D mutation in K-RAS. Similar results were also seen in neurofibroma cell lines that lack functional NF-1, a GTPase for K-RAS. Loss of function of NF-1 leads to constitutive activation of K-RAS. Taken together, these experiments suggest that MLK3 may modulate downstream effectors of K-RAS that are critical for cell proliferation.
The role of MLK3 in pancreatic cancer has not been studied. We tested our hypothesis in well-characterized pancreatic cell lines with known mutations in K-RAS. Both MiaPaCa-2 and PANC-1 cell lines harbor a
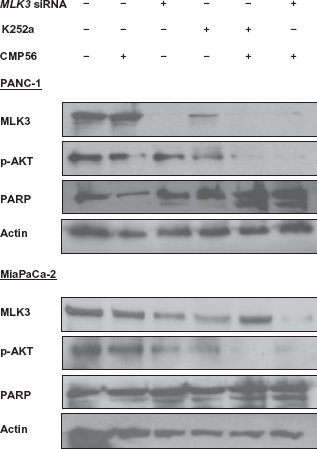
In the current study, we found that MLK3 was over expressed (~3 fold) in both of these pancreatic cancer cell lines compared to the pancreatic epithelial cell line. Silencing of MLK3 by RNA interference in PANC-1 and MiaPaCa-2 cell lines markedly suppressed cell proliferation. As confirmation of this finding, pharmacological inhibition of MLK3 also led to marked suppression of cell proliferation in both pancreatic cancer cell lines. Interestingly, pharmacological inhibition (concentration dependent) led to more significant suppression of cell proliferation than RNA interference. This could suggest that K252a targets other MLK isoforms and that these other MLK isoforms might compensate for silencing of MLK3. It could also be due to off target effects of K252a. K252a also inhibits other protein kinases such as protein kinase C, cyclic nucleotide-dependent protein kinases 32 and Ca2+/calmodulin-dependent protein kinase II. 33 When compared to CEP 1347, an MLK-selective inhibitor, K252a has an essentially similar inhibitory profile towards MLK3. 21 Therefore, we used K252a as the MLK3 inhibitor in our studies. Future studies using MLK3-specific inhibitors are warranted to confirm our results.
To further characterize the downstream molecular events associated with MLK3 inhibition in pancreatic cancer cell lines, additional molecular analyses were performed. In PANC-1 cells, MLK3 silencing led to a decrease in the phosphorylation of JNK, ERK, and AKT. Similar results were seen in the MiaPaCa-2 cell line. Decreased phosphorylation of ERK, JNK and AKT and an increase in apoptosis, as evidenced by significant increase in cleaved caspase-3 activity, was observed upon simultaneous EGFR and MLK3 inhibition (siRNA or K252a). MLK3 inhibition (either by siRNA or K252a) also led to a decrease in the phosphorylation of AKT, which was more robust when MLK3 was inhibited in combination with EGFR inhibition. This could explain the increased effect of the combined drugs on the programmed cell death (apoptosis) in pancreatic cancer. Fleming et al
29
also noticed only 30% apoptotic cell death in MiaPaCa-2 cell line and no apoptosis in PANC-1 cells, when treated with mutant-specific
In conclusion, this study suggests that MLK3 might be an interesting target in pancreatic cancer. Further studies will be necessary to evaluate this possibility. We are in the process of assessing the expression of MLK3 in malignant and benign pancreatic tissue from patients. Preclinical and clinical studies are also needed to study the safety and efficacy of the EGFR and MLK3 inhibitors alone or in combination with other conventional chemotherapeutic agents. If further studies confirm these preliminary findings, then the combination of EGFR and MLK3 inhibitors may be one strategy to improve therapeutic efficacy in treating patients with pancreatic cancer.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.